Executive snapshot
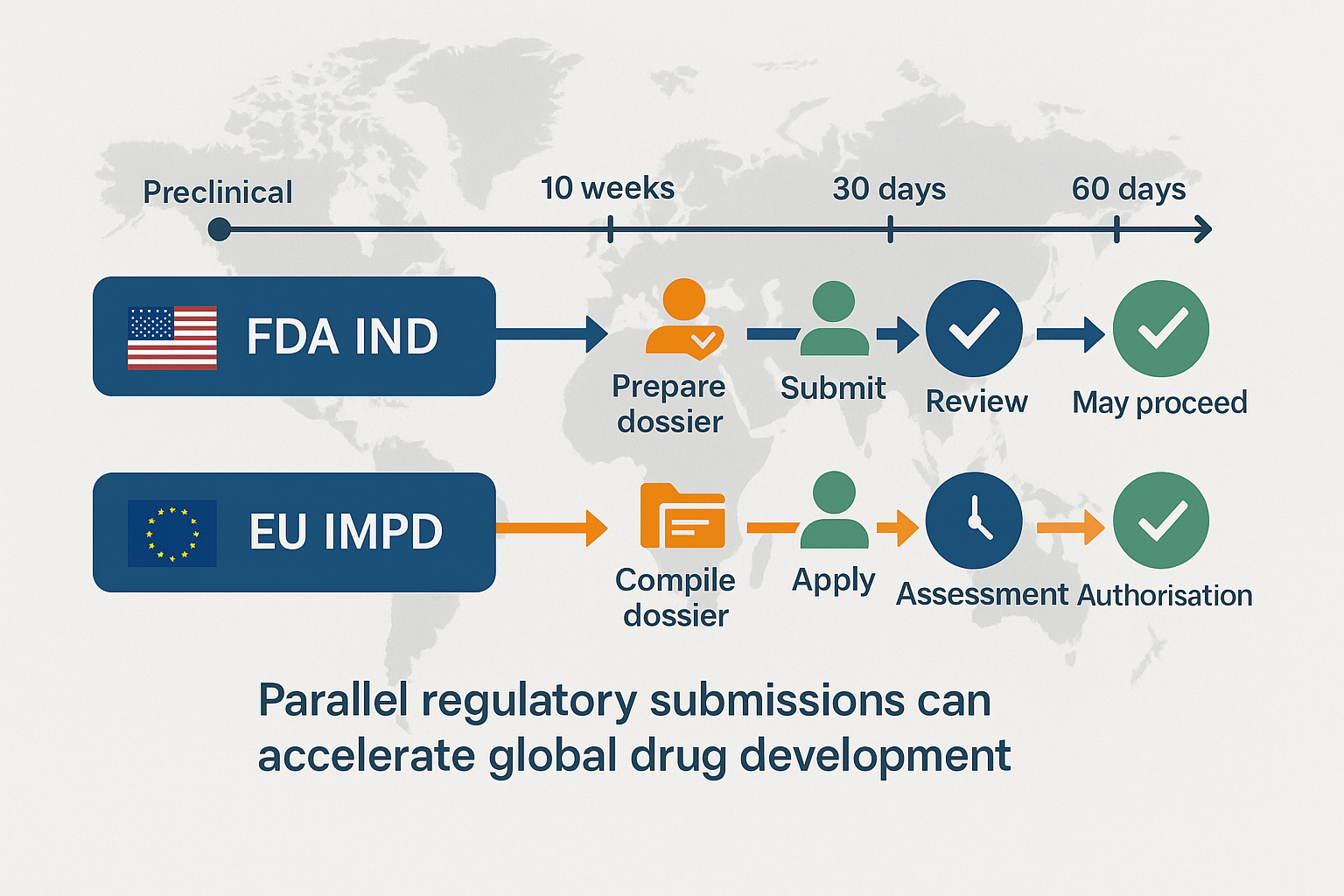
Filing separate first-in-human (FIH) dossiers for the United States and Europe can drag an early-stage program six-plus months behind plan. A harmonised approach—one Common Technical Document (CTD) “master” that slots into both the FDA Investigational New Drug (IND) sequence and the EU Clinical Trials Regulation (CTR) portal (CTIS)—cuts review-cycle queries by ~35 %, lets you open US/EU sites in lock-step, and shows investors a globally scalable package. This article maps the IND and IMPD/CTA frameworks side-by-side, pinpoints six technical gaps that still trip sponsors, and provides a 16-week workflow, case metrics, and ready-to-use check-lists so you can move from pre-clinical data-lock to parallel safe-to-proceed / CTA authorisation in as little as 63 days.
Introduction
Historically, FIH studies followed a sequential path: submit an IND under 21 CFR 312 [1], wait for the FDA’s 30-day review, and then assemble country-specific IMPDs under Regulation (EU) 536/2014 [2]. Today, with investor pressure for parallel site activation and advances in digital publishing, one integrated dossier is feasible—if done correctly from the start.
Despite both systems relying on the CTD structure, regulatory differences remain. The FDA process focuses on a 30-day clinical-hold clock and eCTD format. In contrast, the EU CTR enforces a 60-day joint review (with potential clock-stops) and mandates data publication via CTIS [11]. Below is a comparative overview:
| Key axis | IND | IMPD / CTA |
|---|---|---|
| Non-clinical depth | Two-species, 28-day GLP tox + full ICH S7A/B [8][9] | Selected studies may be waived if justified |
| Starting-dose rationale | Default NOAEL → MRSD [4]; MABEL/PAD optional | EMA FIH Guideline 2017 [3] requires MABEL/PAD only for high-risk assets |
| CMC granularity | High-level Phase 1 specs; stability commitment acceptable | Batch-release specs + QP declaration required up-front |
A harmonised package therefore means adopting the strictest requirement, documenting justified regional deviations, and structuring every file so it drops cleanly into both eCTD and CTIS. Done well, you eliminate duplicate authoring, reduce regulator questions by a third, and keep US/EU Phase 1 timelines in lock-step.
Regulatory foundations — quick reference
| Feature | FDA IND | EU IMPD + CTA |
|---|---|---|
| Governing law | 21 CFR 312 [1] | Regulation (EU) 536/2014 [2] |
| Submission gateway | eCTD (CDER/CBER) | CTIS single portal |
| Clock-stop review | 30-day silent approval unless clinical hold | 60 days + clock-stops for RFIs |
| Primary dossier element | CTD M1-5 | IMPD + admin/ethics annexes |
| Transparency | Confidential | Auto-public disclosure (deferrals possible) [11] |
Where IND & IMPD still diverge—and how to bridge each gap
Non-clinical safety depth
FDA invariably expects two-species 28-day GLP tox; EU may waive selected studies. Bridge: write to the higher bar and attach a one-page waiver memo for EU reviewers.
Starting-dose algorithm
Both regions default to NOAEL → MRSD [4]; EMA’s FIH guideline [3] requires MABEL/PAD only for immune agonists, stimulators, or novel modalities. Bridge: use a single risk-grid slide-deck—page 1 risk table, page 2 MRSD worksheet, page 3 MABEL/PAD if triggers met.
Clinical-pharmacology & DDI package
Small molecules: Follow FDA [5] + ICH M12 [6]; defer transporters if PBPK shows low risk.
mAbs/Proteins: Assess cytokine-mediated CYP effects per FDA 2023 guidance [12].
Peptides/Oligos: Transporter screens if MW <5 kDa [7].
ATMPs: Focus on immunogenicity and conditioning regimen interactions [10].
CMC depth & QP certification
IND allows high-level specs; EU needs batch specs + QP declaration. Bridge: create an IMPD-style spec table and drop the same PDF into IND M3.
Electronic format & transparency
IND eCTD is private; CTIS auto-publishes. Bridge: generate a “public copy” PDF with automated redactions alongside the master file.
Clock-management
FDA clarifying RFIs typically arrive day 14-21; EU RFIs day 40-50. Bridge: a colour-coded gap matrix and a 2-page pre-IND briefing lock the plan before filing.
The single-data-package workflow
| Week | Milestone | Owner | Output |
|---|---|---|---|
| 0-1 | Kick-off audit & gap map | Reg-lead | Airtable tracker |
| 1-2 | Critical-path alignment | PM | Locked Gantt |
| 2-8 | Living gap matrix + modality-flagged DDI table | CP/PK lead | SharePoint “single source” |
| 6 | Compile CTD once; auto-populate IND M1 & IMPD Annex I | Med writer | Draft dossier |
| 8 | CTA submission via CTIS; IND queued | Publisher | eCTD & CTIS packages |
| 6-10 | FDA 30-day review | — | Rapid RFI response |
| 8-16 | EU 60-day review | — | Coordinated RFI response |
| 10-16 | Simultaneous “May Proceed” + CTA authorisations | — | FIH Go |
Common pitfalls & pro tips
| Pitfall | Time lost | Quick fix |
|---|---|---|
| “We’ll localise later.” Two versions emerge. | 4-6 weeks | Write once; track change-log. |
| Country-specific ethics annexes forgotten. | 4 weeks | Annex checklist by Week 4. |
| Stability spec mismatch. | 10-day FDA hold | Use IMPD spec table in IND. |
| CTIS public copy not scrubbed. | Reputation risk | Automated redaction script. |
Frequently asked questions
- Can I file an IMPD verbatim as an IND? No—administrative forms, US-specific REMS text, and eCTD hyperlinks differ.
- How do I handle protocol divergence after approval? Maintain one “master” protocol and track amendments in a single log referenced in both dossiers.
- Does Project Optimus change dose-finding only in the US? No—EMA’s revised oncology guidance is aligned; use the same exposure–response slides.
- Will eCTD v4.0 change CTD structure? Structure stays; v4.0 adds an XML backbone and two-way messaging—tag v3.2.2 sequences now to future-proof.
References
- https://www.ecfr.gov/current/title-21/chapter-I/subchapter-D/part-312
- https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX%3A32014R0536
- https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-strategies-identify-and-mitigate-risks-first-human-and-early-clinical-trials-investigational-medicinal-products-revision-1_en.pdf
- https://www.fda.gov/media/72309/download
- https://www.fda.gov/media/135586/download
- https://database.ich.org/sites/default/files/ICH_M12_Step4_Guideline_2024_0521_0.pdf
- https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revised_en.pdf
- https://database.ich.org/sites/default/files/S7A_Guideline.pdf
- https://database.ich.org/sites/default/files/S7B_Guideline.pdf
- https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-quality-non-clinical-clinical-aspects-medicinal-products-containing-genetically_en.pdf
- https://www.ema.europa.eu/en/human-regulatory/research-development/clinical-trials/clinical-trial-regulation/transparency-rules
- https://www.fda.gov/media/140909/download