When to Use PopPK/PD, Exposure Response, PBPK, and QSP, and When Not To
1. Introduction: Why Quantitative Pharmacology Matters
Model informed development has shifted from optional exploratory analysis to a central part of decision making. Most high value decisions such as target viability, mechanism plausibility, first in human (FIH) dose selection, Phase 2 dose justification, and regulatory interactions now rely on quantitative methods.
Population PK/PD, exposure response (E R), physiologically based PK (PBPK), and quantitative systems pharmacology (QSP) each address a different component of decision uncertainty. The goal is not to use all four techniques, but to apply each one only when it answers a specific development question.
This article summarizes the practical role of these tools, including how they support preclinical development and translational modeling, and situations where they provide more burden than benefit.

2. The Core Quantitative Toolkit
A brief overview:
- Population PK/PD (PopPK/PD): Describes typical PK parameters, variability, covariates, and exposure response relationships.
- Exposure Response (E R): Quantifies how drug exposure relates to efficacy or safety outcomes.
- Physiologically Based PK (PBPK): Mechanistic, whole body modeling using physiology, enzyme and transporter data, and ADME properties.
- Quantitative Systems Pharmacology (QSP): Pathway or network level modeling of drug action, target modulation, and disease dynamics.
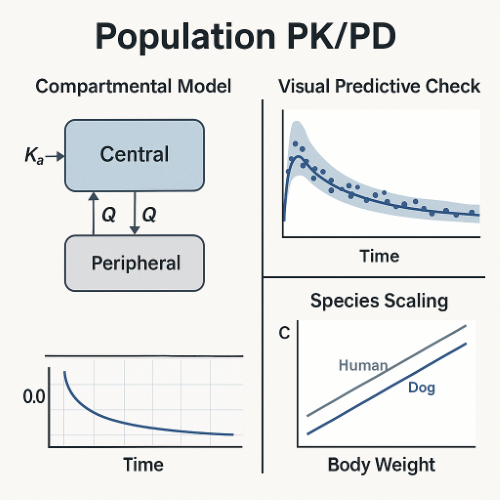
3. Population PK and PopPK PD Models
PopPK is commonly associated with clinical pharmacology, but it also supports translational activities in early development.
Preclinical Use Cases
- Characterizing PK linearity and dose proportionality across species.
- Quantifying in vivo potency using PK PD linkage.
- Capturing delays between exposure and effect.
- Providing stable clearance and volume estimates for allometric scaling.
- Supporting studies with sparse PK designs in larger species.
Clinical Use Cases
- Describing PK variability and covariates.
- Predicting exposures for downstream E R analyses.
- Special population assessments.
- Bridging across formulations or dosing regimens.
When PopPK is Unnecessary
- Early rodent PK programs with minimal variability.
- Linear PK that is easily evaluated using noncompartmental analysis.
- Situations where no decision depends on variability.
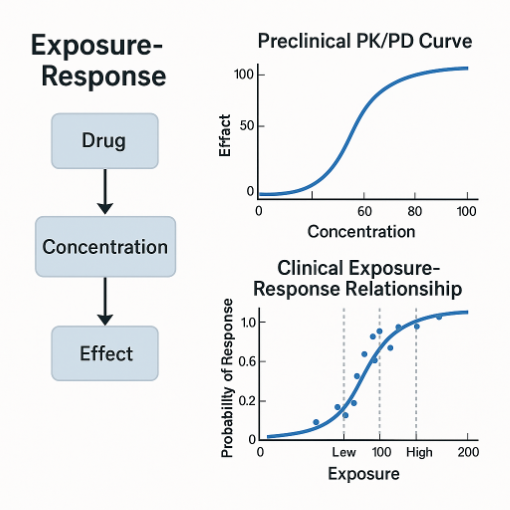
4. Exposure Response (E R) Modeling
Exposure response analysis provides the link between exposure and pharmacology or clinical outcome. This is the foundation of dose selection.
Preclinical Use Cases
- PK PD integration in efficacy and biomarker models.
- Identifying pharmacodynamic drivers such as AUC driven or Cmax driven effects.
- Quantifying EC50, EC90, or target engagement thresholds.
- Translational PK PD modeling to support FIH dose selection.
Clinical Use Cases
- Selecting Phase 2 and Phase 3 doses.
- Quantifying benefit risk tradeoffs.
- Labeling justifications for efficacy and safety endpoints.
- Bridging between formulations or regimens.
When E R is Not Informative
- No meaningful exposure range.
- Clinical endpoints with high noise or rare events.
- Dose selection studies with insufficient differentiation.
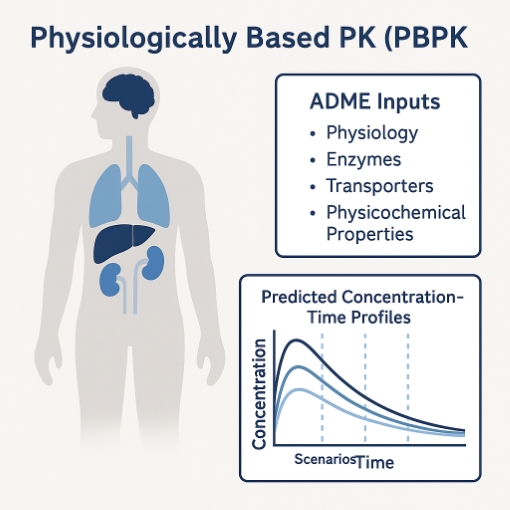
5. PBPK Modeling
PBPK is a powerful option for mechanistic ADME driven questions. It is not required for every modality or for all human PK predictions.
Preclinical Use Cases Where PBPK Adds Value
- IVIVE estimation for human clearance.
- Early evaluation of enzyme and transporter involvement.
- Solubility limited or permeability limited absorption.
- Understanding food effect risks.
- Early formulation strategy development.
- Explaining nonlinearities driven by specific ADME mechanisms.
Clinical Use Cases
- Prospective DDI prediction and potential waiver of clinical studies.
- Organ impairment predictions when mechanisms are understood.
- Pediatric scaling when ontogeny is relevant.
- Transitions between routes of administration.
When PBPK is Unnecessary
- Most monoclonal antibodies with linear PK.
- Small molecules with simple linear PK and no DDI risks.
- When compartmental PK or PopPK with allometry provides adequate answers.
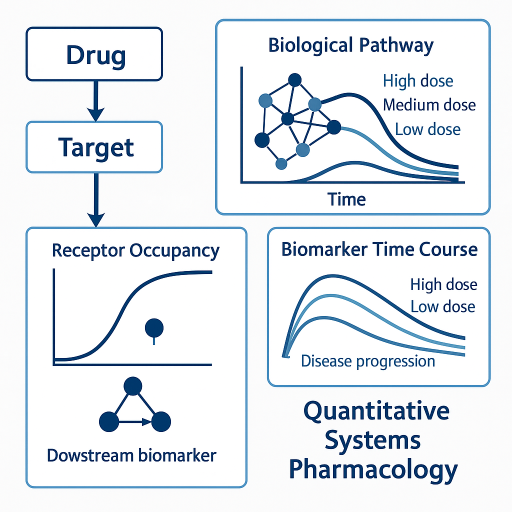
6. Quantitative Systems Pharmacology (QSP)
QSP focuses on biology rather than PK alone. This makes it particularly useful for understanding mechanistic drivers of response.
Preclinical Use Cases
- Target validation and biomarker selection.
- Understanding pathway feedback loops.
- Translational pharmacology for biologics and immune modulators.
- Dose and schedule exploration prior to FIH.
- Connecting in vitro potency with expected in vivo effects.
Clinical Use Cases
- Dose and schedule refinement.
- Interpreting biomarker changes.
- Exploring combination strategies.
- Supporting mechanistic explanations during regulatory interactions.
When QSP is Unnecessary
- Pathways with insufficient data.
- Programs where classical PK PD answers the key question.
- Situations where QSP complexity adds noise rather than insight.
7. Predicting Expected Human PK: Matching the Method to the Modality
Human PK prediction should depend on the modality and the mechanistic complexity of the program.
| Modality | Primary PK Prediction Approach | When This Method Is Appropriate | When to Consider a More Advanced Method |
|---|---|---|---|
| Small molecules | Allometry and compartmental PK | Linear, single pathway clearance; predictable ADME | PBPK for complex ADME, multiple enzymes or transporters, or solubility or permeability limitations |
| Monoclonal antibodies (mAbs) | Allometric scaling of clearance and volume; two compartment PK | Linear PK across species; well understood target binding | Target mediated PK PD modeling for nonlinear TMDD; PBPK rarely needed unless tissue distribution is mechanistically central |
| Bispecifics and T cell engagers | Translational PK PD plus PopPK for each analyte | Multiple analytes with different clearance pathways; strong PD driven effects | PBPK only if tissue uptake or target expression drives distribution |
| ADCs (antibody drug conjugates) | Translational PK PD plus PopPK for total antibody, conjugated drug, and free payload | Distinct analytes with different PK profiles | PBPK for payload distribution or linker related kinetics |
| Radioligands and radiopharmaceuticals | Hybrid compartmental and organ level distribution models | Radiation dosimetry; organ specific uptake | PBPK when uptake mechanisms and tissue kinetics are mechanistic and well characterized |
| Gene therapies | Mechanistic vector and transgene expression models | Capsid distribution and expression driven efficacy | QSP style immune activation or intracellular processing models |
| Cell therapies | Cell expansion and contraction models | Expansion, persistence, and cytokine kinetics | QSP models for immune feedback, cytokine release, and exhaustion dynamics |
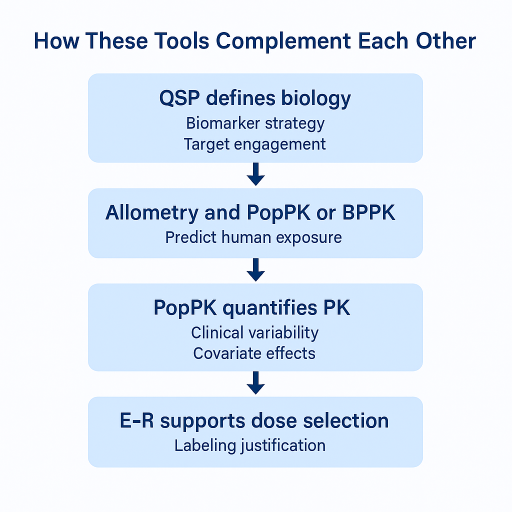
8. How These Tools Complement Each Other
These tools work best as a coordinated framework.
- QSP defines biologic plausibility and key biomarkers.
- Allometry, PBPK, and translational PK PD predict human exposure.
- PopPK quantifies observed PK and sources of variability.
- E R directly supports dose selection and labeling justification.
9. Recommendations for Small and Midsize Biotech Teams
- Choose the modeling technique that answers your specific decision question.
- Match the method to the modality rather than taking a one size fits all approach.
- Integrate modeling early, especially translational PK PD and QSP for novel mechanisms.
- Use PopPK and E R as the foundation for clinical dose justification.
- Avoid building complex PBPK or QSP models when simpler approaches provide the same clarity.
- Maintain a quantitative narrative that connects animal PK PD, predicted human PK, and clinical data.
10. Conclusion
Quantitative pharmacology is most useful when it is applied thoughtfully. PopPK PD, E R, PBPK, and QSP each provide unique insights at different stages of development. When integrated across preclinical and clinical phases, these tools support dose selection, reduce uncertainty, and strengthen regulatory interactions.
The key principle is simple. Use the right tool for the right question. Avoid over modeling, remain grounded in the biology and ADME of the modality, and ensure each analysis supports a real development decision.
How We Can Help
ClinPharm Dev Solutions LLC provides experienced support to help teams determine which quantitative techniques are appropriate for their molecule and their development stage. We assist with tool selection, vendor selection, oversight of outsourced analyses, and hands on execution of PopPK and E R modeling. Our goal is to help you build a clear, defensible quantitative foundation for your program while ensuring that modeling remains decision focused and efficient.
Abbreviations
- ADME: Absorption, distribution, metabolism, excretion
- ADC: Antibody drug conjugate
- AUC: Area under the concentration time curve
- CL: Clearance
- DDI: Drug drug interaction
- EC50, EC90: Concentration producing 50 percent or 90 percent of maximal effect
- E R: Exposure response
- FIH: First in human
- IVIVE: In vitro to in vivo extrapolation
- mAb: Monoclonal antibody
- PBPK: Physiologically based pharmacokinetic modeling
- PD: Pharmacodynamic
- PopPK: Population pharmacokinetic modeling
- PK: Pharmacokinetic
- PK PD: Pharmacokinetic pharmacodynamic modeling
- QSP: Quantitative systems pharmacology
- TMDD: Target mediated drug disposition
- TCE: T cell engager
- VPC: Visual predictive check
References
- FDA. Model Informed Drug Development. Guidance for Industry. 2023.
- EMA. Guideline on the Reporting of Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulation. 2018.
- FDA. Population Pharmacokinetics. Guidance for Industry. 2022.
- Gabrielsson J, Weiner D. Pharmacokinetic and Pharmacodynamic Data Analysis. Fifth Edition.
- Mager D, Jusko W. General pharmacokinetic and pharmacodynamic modeling concepts. Pharmaceutical Research.
- Sorger P et al. Quantitative Systems Pharmacology: An Overview and Clinical Applications.