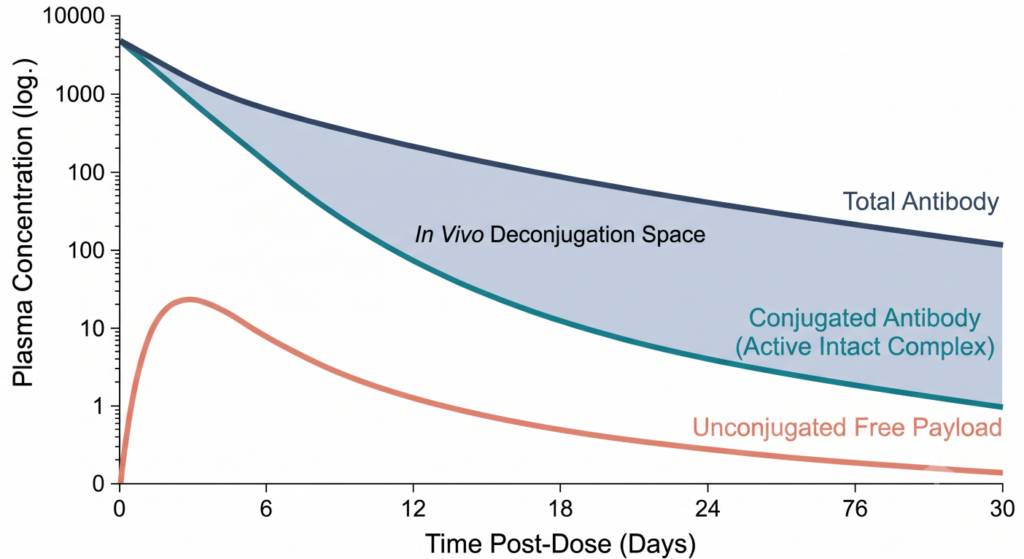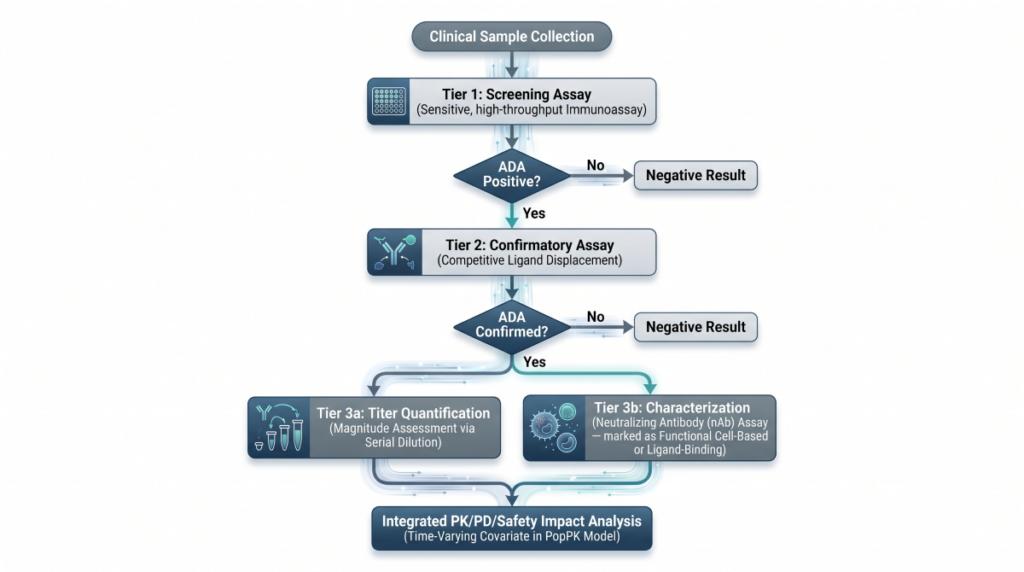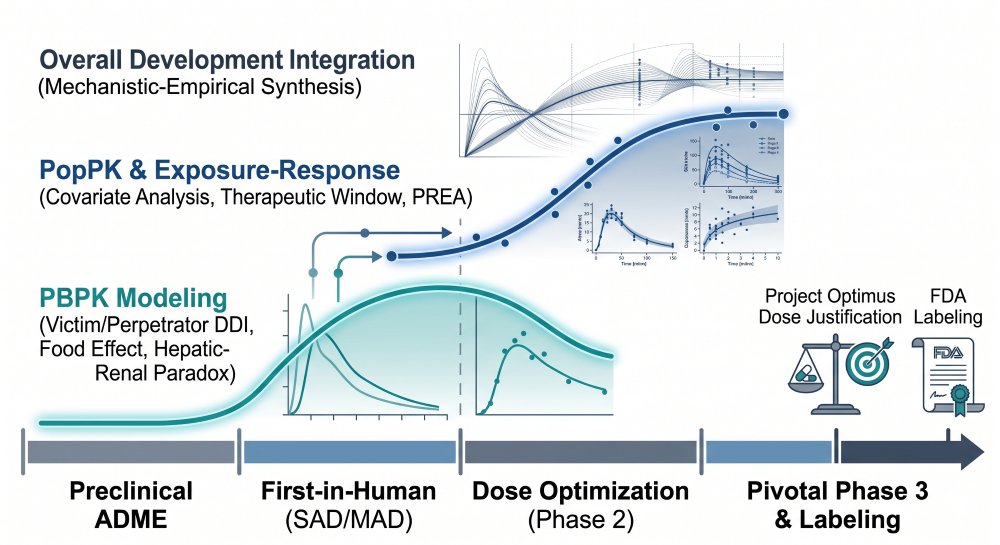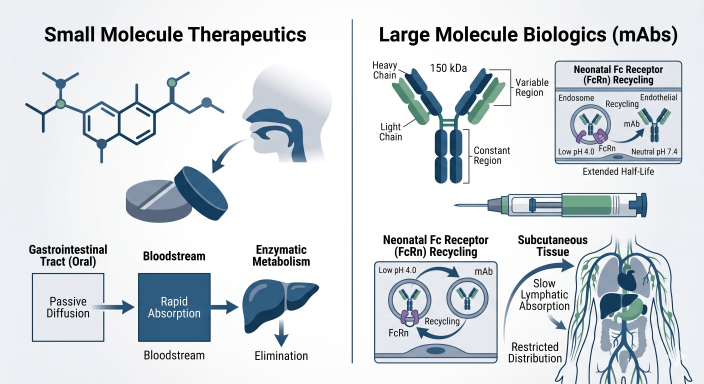
Targeted Radionuclide Therapies (TRTs) and Theranostics: A Quantitative Clinical Pharmacology and DMPK Blueprint
The Modality Matrix Series Portfolio This sixth installment explores the specialized domain of Targeted Radionuclide Therapies (TRTs) and theranostics. Transitioning these dual-modality assets into first-in-human clinical trials requires an advanced quantitative Clinical Pharmacology Plan (CPP) capable of integrating radiation dosimetry, radioactive decay kinetics, and multi-isotope PBPK modeling. Article Sequence Thematic Focus Status Article 1 Overview […]