Strategic Overview: As of April 14, 2026, we are in the critical pre-implementation window for the most significant global pediatric regulatory shifts in a decade. With only weeks remaining until the May 2026 implementation of new incentives in Japan and China, sponsors must reconcile the 24-month “Timing Gap” between EMA and FDA milestones. This analysis provides a transition guide for aligning the EMA PIP and FDA iPSP with the upcoming high-ROI exclusivity “carrots” in Asia.
1. The EMA PIP: Article 16 and the “Absolute Latest” Threshold
In the EU, Regulation (EC) No 1901/2006 dictates a strict submission timeline. Article 16 requires a Paediatric Investigation Plan (PIP) to be filed no later than the completion of human pharmacokinetics (PK) studies in adults—effectively the End of Phase 1.
• The Ideal Window: Submission at End of Phase 1 ensures the PDCO can influence Phase 2 designs. This prevents the “Pediatric Lag” that often delays adult launches.
• The Absolute Latest Threshold: While “duly justified” delays exist, the PDCO generally refuses PIPs submitted after the initiation of confirmatory Phase 3 trials. A late filing at this stage acts as a hard block for the adult Marketing Authorisation Application (MAA) validation.
• Stepwise PIP (sPIP): Now a permanent procedure (as of Feb 2026), the sPIP allows sponsors of innovative therapies to meet the early Article 16 deadline with “conditional milestones,” deferring specific trial designs until Phase 2 data matures.
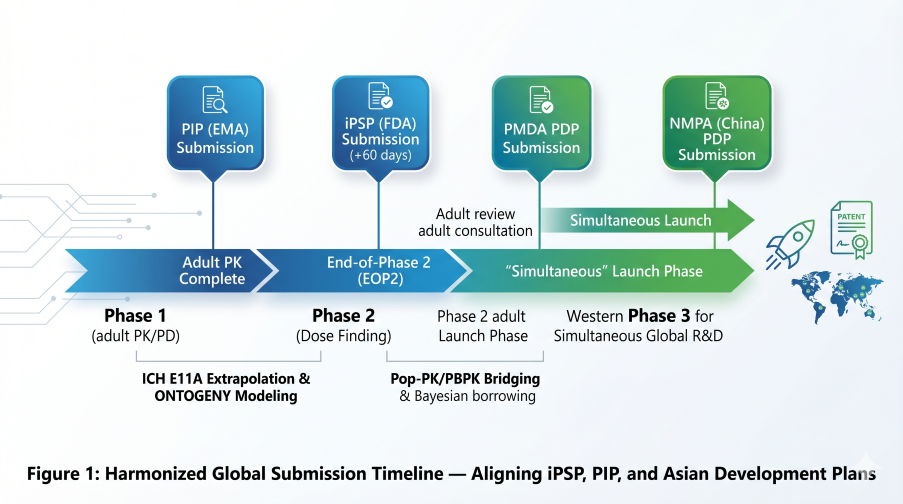
Figure 1: Harmonized Global Submission Timeline — Aligning iPSP, PIP, and upcoming Asian Development Plans
2. The FDA iPSP: Post-EOP2 and the RACE Act Acceleration
The FDA’s initial Pediatric Study Plan (iPSP) is mandated within 60 days of the End-of-Phase 2 (EOP2) meeting. However, current 2026 mandates have accelerated this timeline for specific drug classes:
- Oncology & The RACE Act: Assets targeting molecular mechanisms relevant to pediatric cancers require engagement at the End of Phase 1, effectively synchronizing the iPSP with the EMA PIP timeline.
- Plausible Mechanism Framework (Draft Guidance): For ultra-rare therapies, the FDA is currently moving toward accepting PBPK-anchored mechanistic evidence as a replacement for traditional randomized trials.
3. Technical Matrix: The Global Submission Hierarchy
| Region (Document) | Submission Milestone | Compliance Deadline | 2026 Incentive/Risk |
|---|---|---|---|
| EMA (PIP) | Post-Phase 1 (Adult PK) | Pre-Phase 3 Start | MAA Validation Block |
| FDA (iPSP) | 60 days post-EOP2 | Pre-NDA/BLA Filing | Refusal to File (RTF) |
| PMDA (Japan) | Phase 2 Consultation | End of Adult Review | +12yr Exclusivity (May 1) |
| NMPA (China) | Simultaneous with Western PIP | Post-Phase 3 MRCT | +2yr Protection (May 15) |
4. PMDA (Japan): The 12-Year “Dossier Shield” (Effective May 1, 2026)
Current Status: The MHLW Notifications (issued March 31, 2026) are finalized and set to redefine the re-examination landscape.
Effective May 1, 2026, Japan will officially transition to an extended 12-year re-examination period. This provides a critical Dossier Shield: the PMDA will not accept any generic applications during this window, effectively blocking “skinny labels” of adult versions.
Transition Requirement: To capture the 12-year period, sponsors must confirm their Pediatric Development Plan (PDP) before the adult review period ends. Furthermore, this requires the inclusion of a Japanese Cohort—typically 2–3 sites in a Global MRCT—to provide the necessary ethnic bridging data for local ontogeny validation.
5. NMPA (China): Market Exclusivity & Simultaneous R&D (Effective May 15, 2026)
Current Status: The amended Implementing Regulations of the Drug Administration Law (published Jan 27, 2026) are set for implementation on May 15.
This implementation incentivizes China as a primary-launch market, granting 2 years of standalone market exclusivity for pediatric indications. For the first time, innovators have a defined regulatory mechanism to protect pediatric R&D investment in the region.
The 12-Month Rule: Exclusivity is conditional on Simultaneous R&D. To qualify, sponsors must file in China within 12 months of their first global marketing authorization. Additionally, the NMPA expects local PK Sub-studies within global trials to evaluate metabolic polymorphisms prevalent in Han Chinese populations.
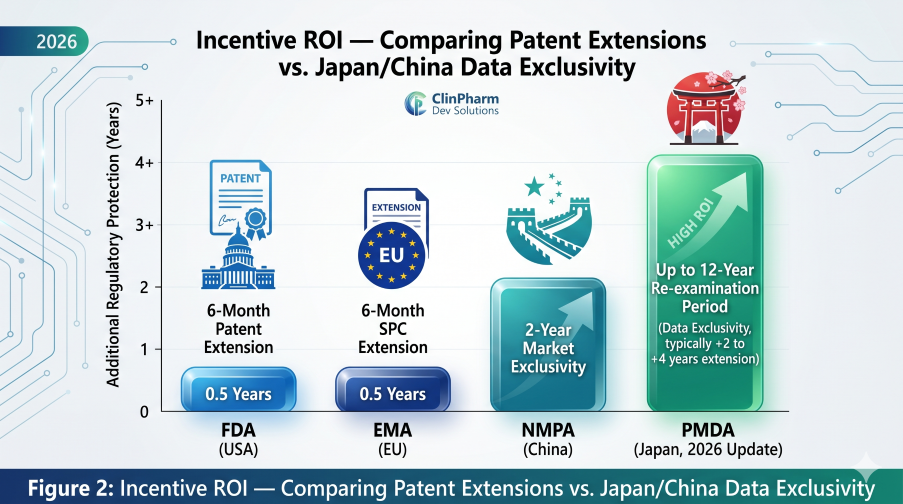
Figure 2: Incentive ROI — Comparing Patent Extensions vs. the upcoming Japan/China Exclusivity Shifts
6. Conclusion: The Pre-Implementation Strategic Pivot
Success in the 2026 landscape requires a single, mechanistic bridge that satisfies all agencies simultaneously. By filing the EMA sPIP at End-of-Phase 1 and leveraging PBPK for Asian ethnic bridging, sponsors can maximize global exclusivity while avoiding the costs of regional silos.
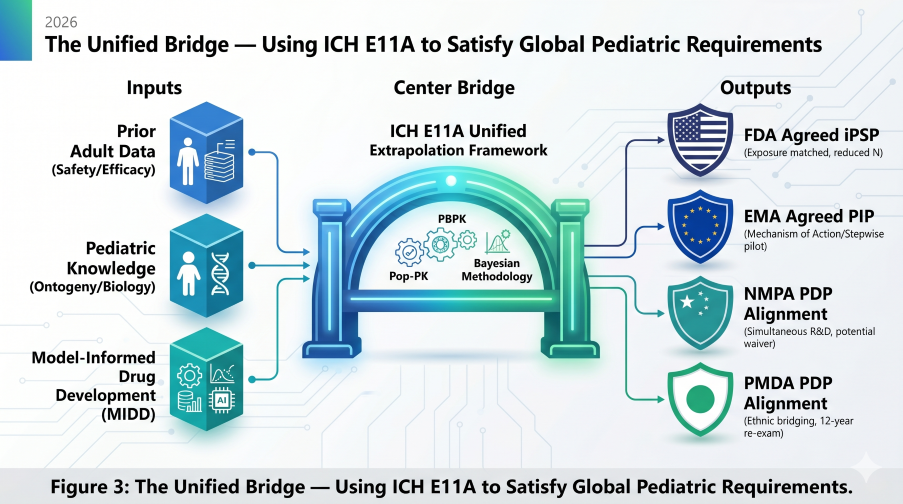
Figure 3: The Unified Bridge — Using ICH E11A to Satisfy Global Pediatric Requirements in 2026
Technical References
- Regulation (EC) No 1901/2006, Article 16 (PIP Submission Timing).
- FDA Draft Guidance (Feb 2026). Plausible Mechanism Framework for Individualized Therapies.
- MHLW Notification (March 31, 2026). 12-Year Re-examination Implementation for Pediatric Drugs.
- NMPA Implementing Regulations (Jan 27, 2026). Pediatric Market Exclusivity and Simultaneous R&D.
- ICH E11A (2024). Pediatric Extrapolation Principles.
Critical Transition FAQ: April/May 2026
Yes, provided you confirm your Pediatric Development Plan (PDP) before the adult review concludes. Per the MHLW Notifications issued on March 31, 2026, the transition window allows for PDP confirmation during the active review cycle to secure the 12-year shield.
Generally, no. The 2-year market exclusivity incentive outlined in the Jan 27, 2026 Implementing Regulations is designed for new registration applications filing after the May 15 implementation. For currently marketed drugs, the NMPA continues to utilize priority review pathways without the specific 2-year market block.
No. It is a voluntary procedure, but it is highly recommended for innovative ATMPs or drugs targeting rare pediatric diseases. It allows you to satisfy the legal “End of Phase 1” deadline while maintaining the flexibility to adjust trial designs as adult efficacy data emerges.