Executive Summary
On March 18, 2026, the FDA issued a landmark draft guidance on New Approach Methodologies (NAMs), signaling a transformative shift in the preclinical landscape. For small biotechs, the guidance provides a formal pathway to use in silico models, organ-on-a-chip technology, and other non-animal methods to support regulatory decision-making. This is no longer just an ethical preference; it is a strategic maneuver to bypass animal study bottlenecks, reduce capital burn, and accelerate the path to IND through human-relevant data.
1. The 2026 Paradigm Shift: From Animals to Algorithms
For decades, animal toxicology was the standard “gatekeeper” for clinical trials. However, with the FDA Modernization Act 2.0 and the newly released March 2026 draft guidance [1], the Agency has formalized how sponsors can use NAMs as primary evidence. The focus has shifted from “species-matching” to Human Biological Relevance.
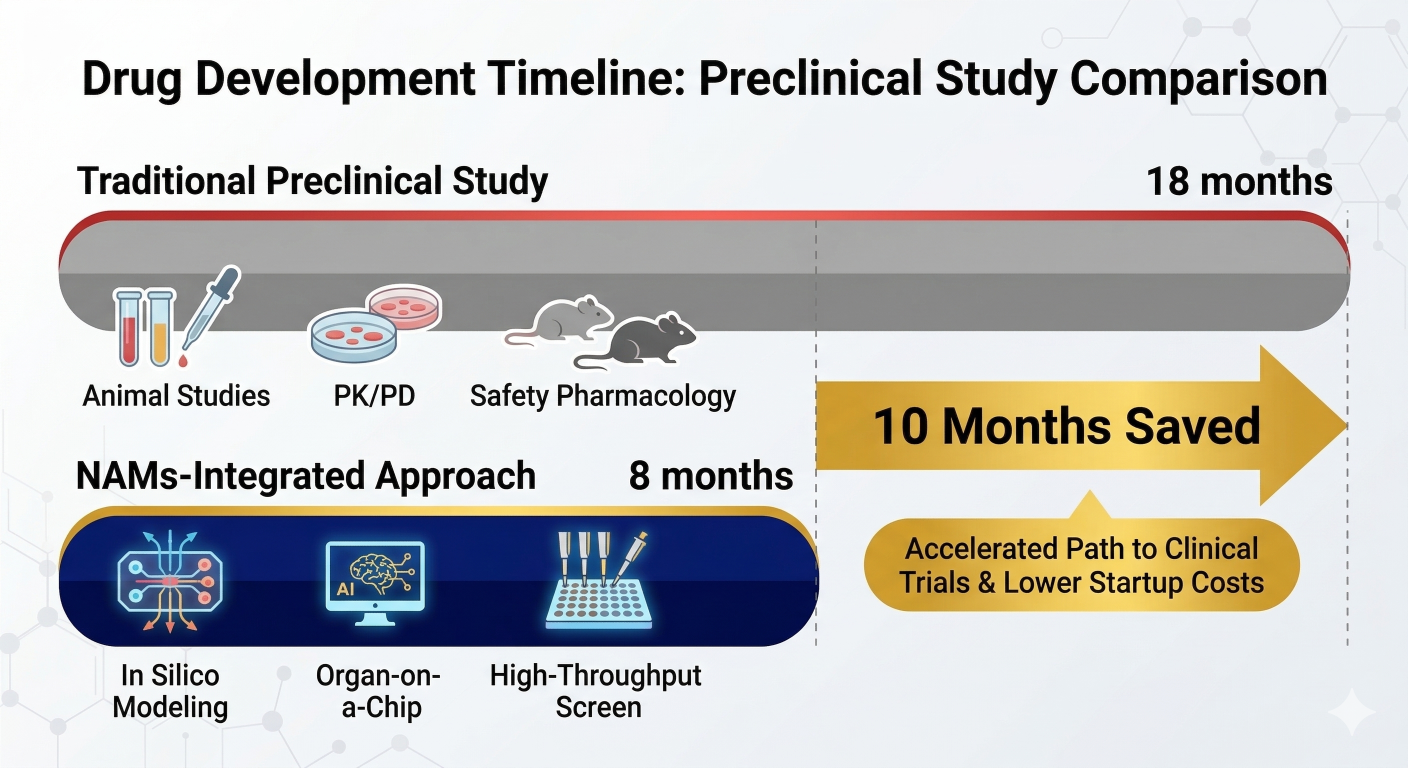 Figure 1: Strategic Acceleration. Leveraging NAMs-Integrated workflows can reduce early-stage safety assessment timelines by up to 60%.
Figure 1: Strategic Acceleration. Leveraging NAMs-Integrated workflows can reduce early-stage safety assessment timelines by up to 60%.
2. Defining “Fit-for-Purpose” (FFP) Validation
The regulatory hurdle for NAMs isn’t “perfection,” but “suitability.” The 2026 guidance emphasizes that a model must be Fit-for-Purpose for the specific decision being made. If you are using an in silico tool to predict a specific metabolic liability or a cardiotoxic risk, you only need to validate the model for that specific mechanism [1, 2].
| Requirement | Traditional Animal Tox | NAMs-Integrated (The 2026 Standard) |
|---|---|---|
| Primary Evidence | In vivo multi-species studies | Validated in silico/in vitro systems |
| Predictivity | Species-specific (Cross-species gaps) | Human-relevant (Mechanism-specific) |
| Typical Timeline | 12 – 18 Months | 4 – 7 Months |
| Startup Advantage | High CapEx / Low Agility | Operational Agility / Targeted Burn |
Case Study: Bypassing the NHP Bottleneck
In late 2025, a small oncology startup developing a T-cell engager faced a 14-month delay for Non-Human Primate (NHP) slots. By leveraging the principles now formalized in the March 2026 guidance, the sponsor integrated a QSP (Quantitative Systems Pharmacology) model with human-on-a-chip cytokine release assays. The FDA accepted this “weight of evidence” package to justify a safe First-in-Human starting dose, allowing the sponsor to skip the NHP study entirely and move into Phase I nearly a year ahead of schedule [4].
3. The Four Technical Pillars of NAMs Acceptance
To succeed with a NAMs-based submission in 2026, the FDA looks for four technical cornerstones:
- Context of Use (COU): A clear statement of the specific regulatory decision the model is intended to support.
- Biological Relevance: Proof that the system captures the human physiological response or target interaction.
- Technical Robustness: Evidence of reproducibility across different batches and laboratories.
- Transparency: Full access to the computational logic and assumptions (the end of “Black Box” modeling).
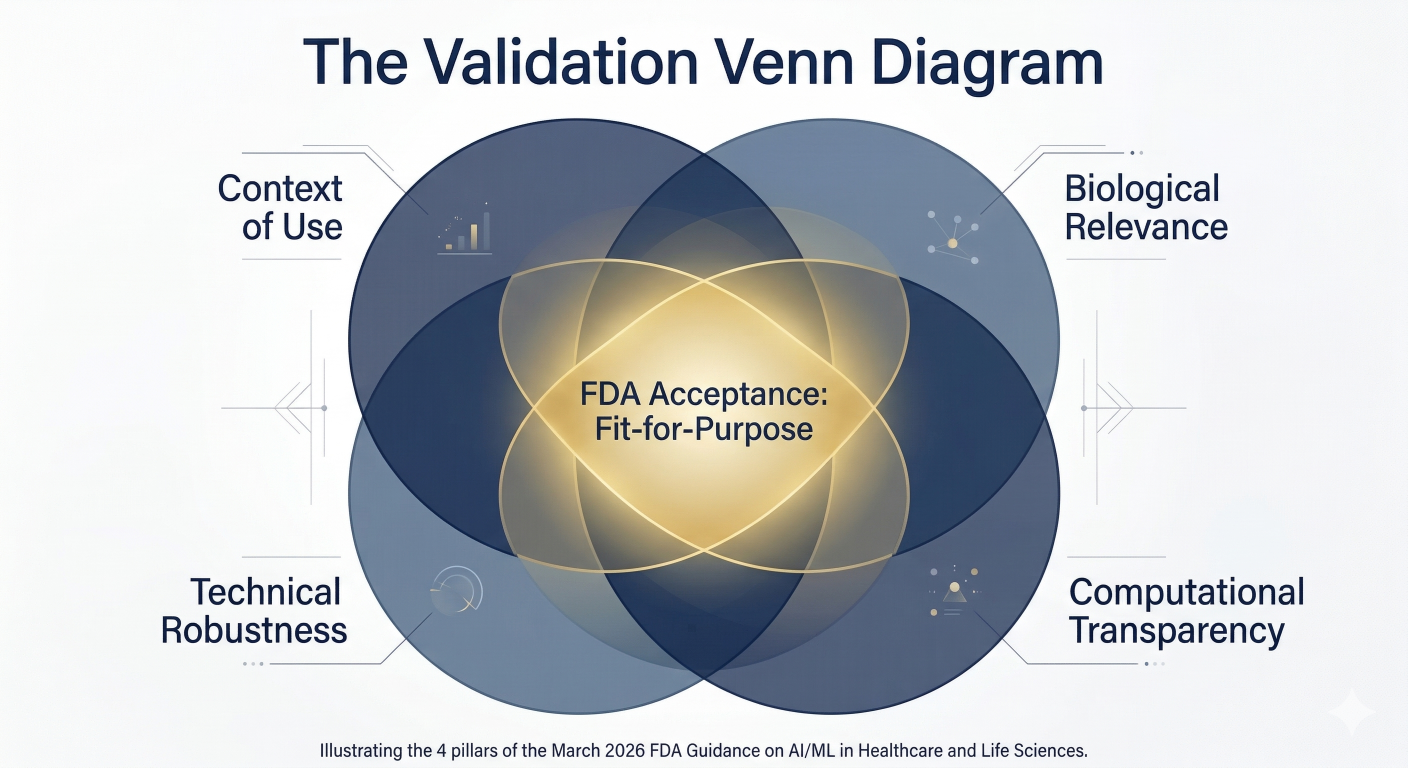 Figure 2: The Validation Framework. The three pillars required to move a NAM from supportive data to primary regulatory evidence.
Figure 2: The Validation Framework. The three pillars required to move a NAM from supportive data to primary regulatory evidence.
Conclusion: Strategic Agility in the Post-Animal Era
The FDA’s March 2026 NAMs guidance is a game-changer for lean biotech organizations. It shifts the competitive advantage from those with the largest animal facilities to those with the most sophisticated Pharmacometrics and DMPK strategies. Navigating this “Fit-for-Purpose” pathway requires a deep technical understanding of both the science and the evolving regulatory nuance. At ClinPharm Dev Solutions, we ensure your science is translated into the language the FDA expects to hear.
Is Your Program a Candidate for NAMs?
Don’t let animal study delays stall your IND. Let us evaluate if your safety package can be accelerated using New Approach Methodologies.
Schedule a Strategic AssessmentAbbreviations
- COU: Context of Use
- DMPK: Drug Metabolism and Pharmacokinetics
- FDA: Food and Drug Administration
- FFP: Fit-for-Purpose
- NAMs: New Approach Methodologies
- PBPK: Physiologically Based Pharmacokinetics
- QSP: Quantitative Systems Pharmacology
References
- FDA Draft Guidance (March 18, 2026). General Considerations for the Use of New Approach Methodologies in Drug Development.
- ICH S12 (2025). Technical and Regulatory Considerations for In Silico Pharmacology.
- Marshall, S. et al. (2025). The Economics of NAMs: Accelerating Startup Timelines. CPT: PSP.
- FDA Case Study Repository (2026). Alternative Safety Assessments for Biologics.
Expert FAQ: FDA March 2026 NAMs Guidance
1. What is the primary objective of the FDA’s March 2026 NAMs draft guidance?The primary objective is to formalize a regulatory framework for using New Approach Methodologies (NAMs), such as in silico models and in vitro microphysiological systems, to support or replace traditional animal testing in preclinical safety assessments. It provides a roadmap for sponsors to validate these systems through a “Fit-for-Purpose” framework.
2. How does the FDA define “Fit-for-Purpose” (FFP) validation for NAMs?
Fit-for-Purpose (FFP) validation means that the NAM must be validated specifically for its intended context of use (COU). This involves demonstrating that the model—whether it is an Organ-on-a-Chip or a QSAR model—is robust, reproducible, and provides human-relevant biological data that can reliably inform a specific regulatory decision.
3. What are the “Four Pillars of Validation” required for NAMs submissions?
The four pillars required by the FDA include: Biological Relevance, Technical Robustness, Context of Use, and Analytical Validation.
4. Can NAMs be used to waive traditional IND-enabling animal studies?
Yes. Under the 2026 guidance, the FDA allows for “Regulatory Biowaivers” if a sponsor can prove that a validated NAM provides equivalent or superior safety data.
5. How should sponsors approach the FDA regarding a NAM-based strategy?
Sponsors should engage the FDA early, ideally during a Pre-IND (PIND) meeting, to align on the validation plan before the formal submission.