The Modality Matrix Series Portfolio
This third report shifts focus from low-molecular-weight chemical entities to engineered macromolecular biologics. The clinical pharmacology plan (CPP) for large molecules requires multi-dimensional characterization of tissue convection, target-mediated saturation, spatial binding kinetics, and immunogenicity profiles to clear modern regulatory barriers.
| Article Sequence | Thematic Focus | Status |
|---|---|---|
| Article 1 | Overview of Modalities and the Clinical Pharmacology Plan | Published |
| Article 2 | Small Molecule PK and Bioavailability | Published |
| Article 3 | Large Molecule Biologics and Subcutaneous Delivery | Current Report |
Introduction: The Physics of Biologic Macromolecules
The transition from small molecule chemical entities to engineered large molecule biologics introduces entirely distinct pharmacokinetic and pharmacodynamic paradigms. Unlike small molecules, which are governed by passive diffusion and phase-based enzymatic metabolism, large molecules are subject to convective tissue transport, neonatal Fc receptor recycling, and target-mediated saturation [3]. Modern regulatory frameworks have displaced empirical dose selection for biologics, mandating instead a mechanistic quantification of target engagement and fluid-phase kinetics. The FDA and EMA demand a highly mechanistic Clinical Pharmacology Plan (CPP) that characterizes the spatial and temporal interaction between the macromolecule and its biological targets.
This third report in “The Modality Matrix” series details the essential components of a large molecule CPP. It focuses predominantly on monoclonal antibodies (mAbs) and bispecific antibodies (bsAbs), outlining the strategies required to manage absorption barriers, nonlinear clearance, dual-target engagement kinetics, and immunogenicity.
Section 1: Absorption Barriers and Subcutaneous Delivery Optimization
While intravenous (IV) administration ensures complete systemic bioavailability, patient-centric drug development has driven a widespread shift toward subcutaneous (SC) delivery. Integrating an SC formulation into the CPP introduces significant physiological complexities that must be addressed early in development.
Due to their high molecular weight, intact antibodies cannot cross the vascular endothelium directly from the subcutaneous space via passive diffusion. Instead, they must be absorbed via the lymphatic system, a convective transport process driven by interstitial fluid pressure and lymphatic contractions [3]. This results in a prolonged absorption phase, with typical maximum plasma concentrations (Cmax) occurring 2 to 8 days post-dose.
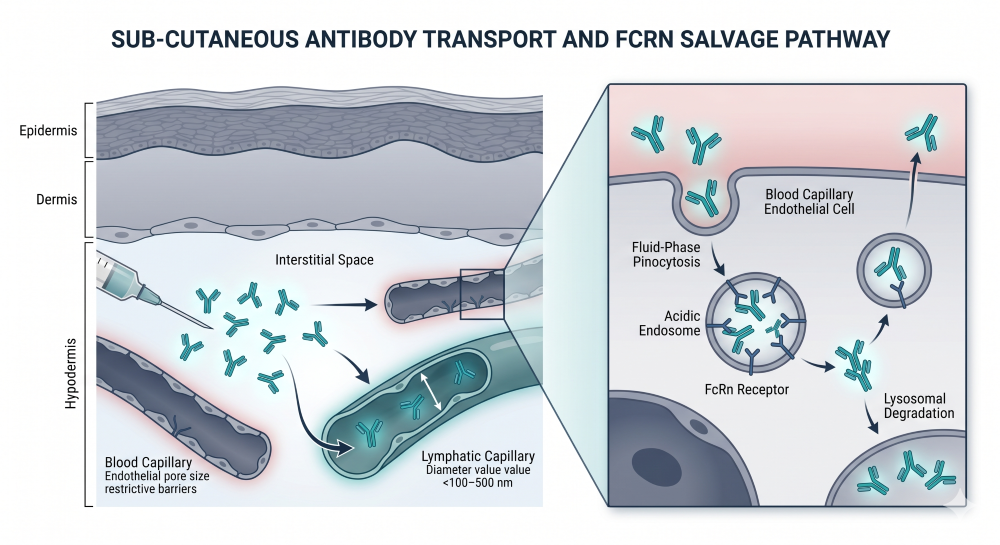
Figure 1: Subcutaneous Absorption and Lymphatic Convection Architecture
Furthermore, absolute SC bioavailability is typically incomplete, ranging from 50% to 80% due to presystemic catabolism by proteolytic enzymes within the hypodermis. The CPP must outline a rigorous bridging strategy to demonstrate that the SC regimen achieves non-inferior systemic exposure, measured as the Area Under the Curve (AUC), and maintains trough concentrations (Ctrough) equal to or greater than those established by the IV regimen.
Section 2: Target-Mediated Drug Disposition (TMDD) and Nonlinear Clearance
The systemic elimination of therapeutic antibodies is characterized by the coexistence of two distinct pathways: a parallel linear, non-specific clearance mechanism and a nonlinear, target-mediated clearance mechanism [4].
The linear pathway occurs via fluid-phase pinocytosis by vascular endothelial cells, followed by lysosomal degradation. IgG-based antibodies are uniquely protected from this pathway by the neonatal Fc receptor (FcRn), which binds to the antibody within the acidic endosome and recycles it back into the systemic circulation, extending the typical elimination half-life (t1/2) to approximately 14 to 21 days.
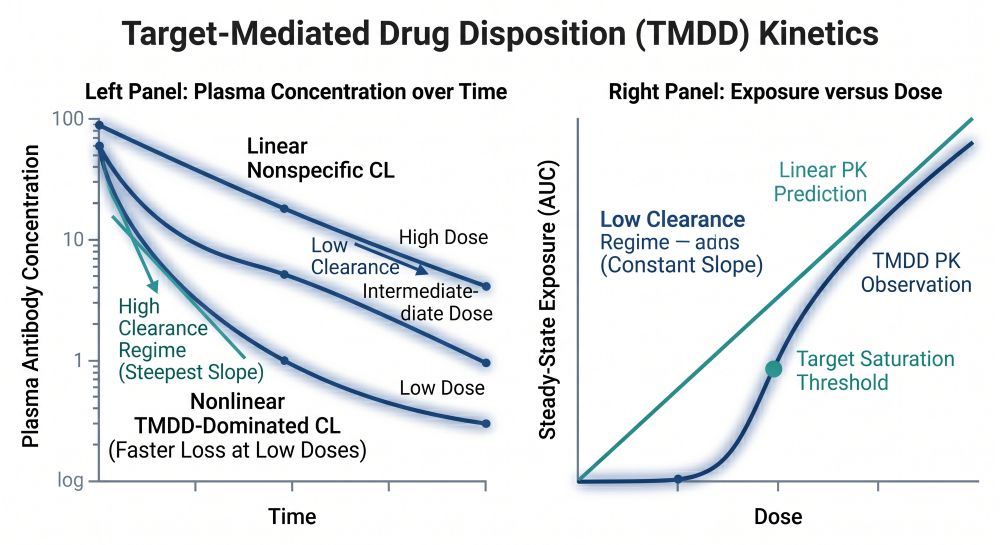
Figure 2: The TMDD Saturation Profile Across Ascending Dose Tiers
The nonlinear pathway is driven by Target-Mediated Drug Disposition (TMDD). This occurs when the antibody binds with high affinity to its specific pharmacological target on the cell surface, triggering receptor-mediated endocytosis and rapid cellular degradation [4]. At low doses, clearance is dominated by this highly efficient, saturable TMDD pathway, causing a non-proportional increase in exposure as doses ascend. At high doses, the target receptors become completely saturated, and clearance normalizes to the slower, linear FcRn-mediated pathway.
In oncology CPPs, the plan must account for time-varying clearance. Population pharmacokinetic analyses frequently demonstrate that mAb clearance decreases over the duration of treatment. This shift is a direct reflection of target baseline dynamics: as the therapeutic antibody successfully reduces tumor burden, the total target pool diminishes, systematically shrinking the contribution of the nonlinear TMDD pathway over time [4].
Section 3: Bispecific Antibodies and Spatial Pharmacometrics
Bispecific antibodies (bsAbs) incorporate dual-targeting architectures, allowing a single molecule to simultaneously engage two distinct antigens [2]. This capability enables novel mechanisms of action, such as T-cell redirection, where one arm of the bsAb binds to a tumor-associated antigen (TAA) and the other binds to an activating receptor on an immune effector cell, such as CD3 on T-cells.
The pharmacology of a T-cell redirecting bsAb is governed by spatial trimeric complex kinetics. Efficacy is not driven simply by systemic exposure, but by the physical formation of a bridge consisting of the tumor cell, the bispecific antibody, and the effector T-cell [5]. The CPP must utilize sophisticated pharmacometric models to predict and avoid the “hook effect,” which manifests as a bell-shaped dose-response curve. At excessively high systemic concentrations, the individual arms of the bsAb saturate both the tumor antigens and the CD3 receptors independently, which prevents the required cellular cross-linking and abolishes functional pharmacological activity despite high drug exposure.
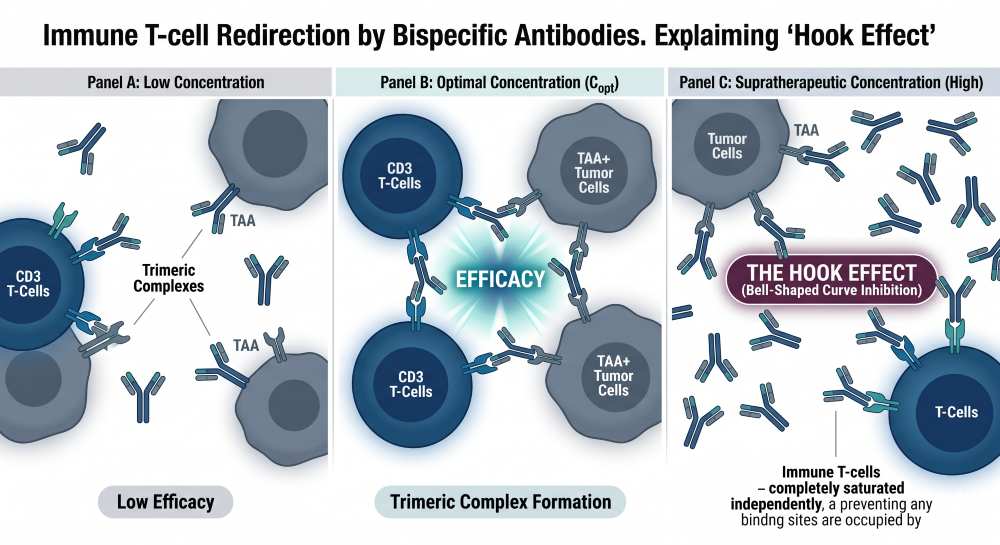
Figure 3: Trimeric Complex Kinetics and Hook Effect Synapse Visualization
Furthermore, initial First-in-Human (FIH) dose selection for bsAbs requires extreme caution due to the risk of hyper-acute immune activation, such as cytokine release syndrome. The CPP must abandon the traditional No Observed Adverse Effect Level (NOAEL) approach in favor of a Minimum Anticipated Biological Effect Level (MABEL) strategy [2]. The MABEL calculation integrates in vitro receptor occupancy models and cell-based functional assays to select a conservative starting dose based on the lowest concentration required to trigger a measurable pharmacological response.
Section 4: Immunogenicity and Anti-Drug Antibody (ADA) Dynamics
Because large molecules are complex biological products, they possess an inherent potential to provoke an immune response in human subjects. The development of Anti-Drug Antibodies (ADAs) can profoundly alter the drug’s safety and efficacy profile, making a multi-tiered immunogenicity assessment a mandatory component of the CPP [1].
The bioanalytical strategy outlined in the CPP follows a structured screening and confirmatory cascade:
- Screening Assay: An initial highly sensitive immunoassay detects any binding antibodies within the clinical matrix.
- Confirmatory Assay: Samples testing positive are subjected to competitive displacement to confirm true antigen specificity.
- Titer Quantification: Confirmed positive samples are serially diluted to determine the magnitude of the immune response.
- Neutralizing Assay (nAb): Confirmed positive samples are evaluated in functional cell-based or competitive ligand-binding assays to determine if the ADAs physically block the antibody’s target-binding domain.
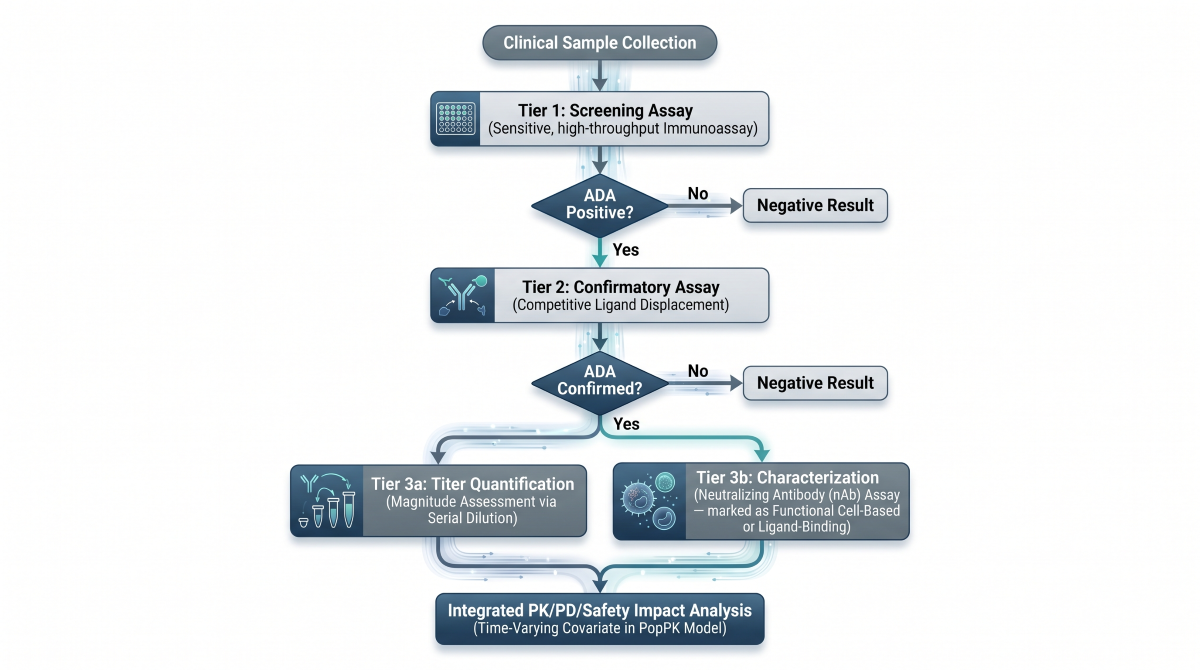
Figure 4: Multi-Tiered Bioanalytical Immunogenicity Assessment Cascade
The CPP must integrate ADA status as a time-varying covariate within the population pharmacokinetic model [1]. Sustained, high-titer ADAs frequently accelerate clearance by forming immune complexes that are rapidly cleared by the mononuclear phagocyte system, drastically reducing the drug’s AUC and clinical efficacy. Conversely, neutralizing antibodies can completely neutralize functional activity without immediately altering total systemic concentrations.
Section 5: The MIDD Engine for Biologics
Modeling and Simulation (M&S) for large molecules shifts focus away from the liver-enzyme parameters used for small molecules, moving instead toward physiological fluid dynamics and target expression levels.
- Minimal Physiologically Based Pharmacokinetics (mPBPK): Unlike small molecule full-body PBPK, biologics utilize mPBPK models that simplify body tissues into leaky or tight vascular compartments. These models use lymph-to-plasma capillary reflection coefficients to simulate convective tissue distribution and predict target site exposure [5].
- Population Pharmacokinetics (PopPK): PopPK models for biologics focus heavily on physiological covariates. Body weight is almost universally a significant covariate for volume of distribution (Vc and Vp) and linear clearance (CL), justifying the transition from flat dosing to weight-based or body-surface-area-based dosing regimens.
- Quantitative Systems Pharmacology (QSP): QSP models are heavily deployed for bispecific antibodies [5]. By mathematically integrating target shedding, receptor internalization rates, and cellular trafficking patterns, QSP models simulate the exact spatial microenvironment of the tumor synapse, optimizing dose scheduling to maximize trimeric complex formation while minimizing systemic toxicity.
Comparative Clinical Pharmacology of mAbs and Bispecifics
| Parameter | Monoclonal Antibodies (mAbs) | Bispecific Antibodies (bsAbs) |
|---|---|---|
| Target Binding Architecture | Monovalent or bivalent affinity toward a single antigen epitope. | Dual specificity, binding two distinct antigen epitopes simultaneously. |
| Elimination Clearance Pathways | Driven by non-specific pinocytosis (linear) and target-mediated endocytosis (nonlinear TMDD). | Heavily dominated by multi-target TMDD, complex internalizing receptors, and rapid immune-cell mediated turnover. |
| FIH Dosing Strategy | Typically justified via traditional scaling or standard NOAEL approaches. | Strictly mandated to use the conservative MABEL approach based on in vitro functional sensitivity. |
| Dose-Response Dynamics | Monotonic or sigmoidal response curves where increasing exposure correlates with increased effect. | Non-monotonic, bell-shaped response profiles driven by the hook effect at saturating concentrations. |
| Immunogenicity Liability | Low to moderate risk, dependent on humanization and target location. | Significantly elevated risk due to structural linkers, novel neo-epitopes, and intense immune cell engagement. |
| Primary M&S Tools | Standard PopPK covariate analysis paired with empirical TMDD equations. | Mechanistic QSP modeling and multi-compartment trimeric complex binding equations. |
Conclusion
The Clinical Pharmacology Plan for large molecules requires a comprehensive understanding of cellular physiology and macromolecular architecture. By transitioning away from empirical playbooks and deploying advanced pharmacometric tools like mPBPK and QSP modeling, clinical pharmacologists can accurately quantify the dynamics of TMDD, optimize subcutaneous bioavailability, manage immunogenicity liabilities, and safely navigate the delicate spatial requirements of bispecific therapies. This disciplined approach eliminates regulatory ambiguity and ensures the rapid development of safe, highly optimized biological assets.
Abbreviations
ADA: Anti-Drug Antibody
AUC: Area Under the Curve
bsAb: Bispecific Antibody
CL: Clearance
Cmax: Maximum Concentration
CPP: Clinical Pharmacology Plan
Ctrough: Trough Concentration
FcRn: Neonatal Fc Receptor
FIH: First-in-Human
IV: Intravenous
mAb: Monoclonal Antibody
MABEL: Minimum Anticipated Biological Effect Level
MIDD: Model-Informed Drug Development
mPBPK: Minimal Physiologically Based Pharmacokinetic
nAb: Neutralizing Antibody
NOAEL: No Observed Adverse Effect Level
PopPK: Population Pharmacokinetics
QSP: Quantitative Systems Pharmacology
SC: Subcutaneous
TAA: Tumor-Associated Antigen
TMDD: Target-Mediated Drug Disposition
Technical References
- FDA Guidance for Industry (2025). Immunogenicity Assessment for Therapeutic Proteins.
- FDA Guidance for Industry (2026). Clinical Pharmacology Considerations for Antibody-Based Therapeutics with Multi-Targeting Architectures.
- EMA Guideline (2024). Development of Monoclonal Antibodies and Derivatives: Clinical and Non-Clinical Aspects.
- Mager, D. E., & Jusko, W. J. (2001). General pharmacokinetic model for target-mediated drug disposition. Journal of Pharmacokinetics and Pharmacodynamics.
- Koch, G., et al. (2023). Modeling and simulation of bispecific antibodies: A review of mathematical concepts and applications. CPT: Pharmacometrics & Systems Pharmacology.