The Modality Matrix Series Portfolio
This fourth installment bridges the gap between small and large molecule pharmacokinetics by examining engineered multi-component systems. Managing Antibody-Drug Conjugates (ADCs) requires an integrated Clinical Pharmacology Plan (CPP) designed to align with the newly finalized 2026 ICH M15 model credibility standards.
| Article Sequence | Thematic Focus | Status |
|---|---|---|
| Article 1 | Overview of Modalities and the Clinical Pharmacology Plan | Published |
| Article 2 | Small Molecule PK and Bioavailability | Published |
| Article 3 | Large Molecule Biologics and Subcutaneous Delivery | Published |
| Article 4 | Antibody-Drug Conjugates (ADCs) and Multi-Analyte PK | Current Report |
Introduction: The Ultimate Structural Hybrid
Antibody-Drug Conjugates (ADCs) represent a sophisticated architectural paradigm in precision oncology. They are engineered to combine the high target selectivity of monoclonal antibodies (mAbs) with the intense anti-proliferative potency of a small molecule cytotoxic payload. By covalently tethering these cytotoxic agents via a specialized chemical linker, the systemic exposure, and subsequent off-target toxicity, of payloads too toxic for traditional systemic administration can be minimized.
However, this multi-component assembly presents unique pharmacokinetic (PK) and pharmacodynamic (PD) profiles that confound standard single-modality evaluation frameworks. In the modern regulatory landscape defined by the FDA’s Project Optimus initiative and the finalized ICH M15 standards, empirical dose-finding is obsolete. An ADC asset requires a robust Clinical Pharmacology Plan (CPP) focused on the mechanistic quantification of structural deconjugation, multi-analyte clearance profiles, payload-driven extrinsic drug interaction liabilities, and spatial tumor microenvironment dynamics.
Section 1: The Three-Analyte PK Paradox and Bioanalytical Characterization
The core operational complexity of an ADC clinical pharmacology strategy lies in tracking multiple distinct structural entities concurrently. Unlike conventional modalities, evaluating an ADC requires three separate, validated bioanalytical assays to fully map systemic disposition:
- Total Antibody: Measures the concentration of all circulating antibody backbones, irrespective of whether they remain conjugated to the payload or are fully deconjugated.
- Conjugated Antibody (Active Intact Complex): Quantifies the concentration of antibody backbones carrying at least one active payload molecule. This analyte reflects the primary targeted delivery vector.
- Unconjugated Free Payload: Tracks the systemic concentration of the small molecule cytotoxin (such as monomethyl auristatin E [MMAE], DXd, or exatecan derivatives) that has been cleaved from the antibody via premature systemic deconjugation or leaked out of target cell boundaries.
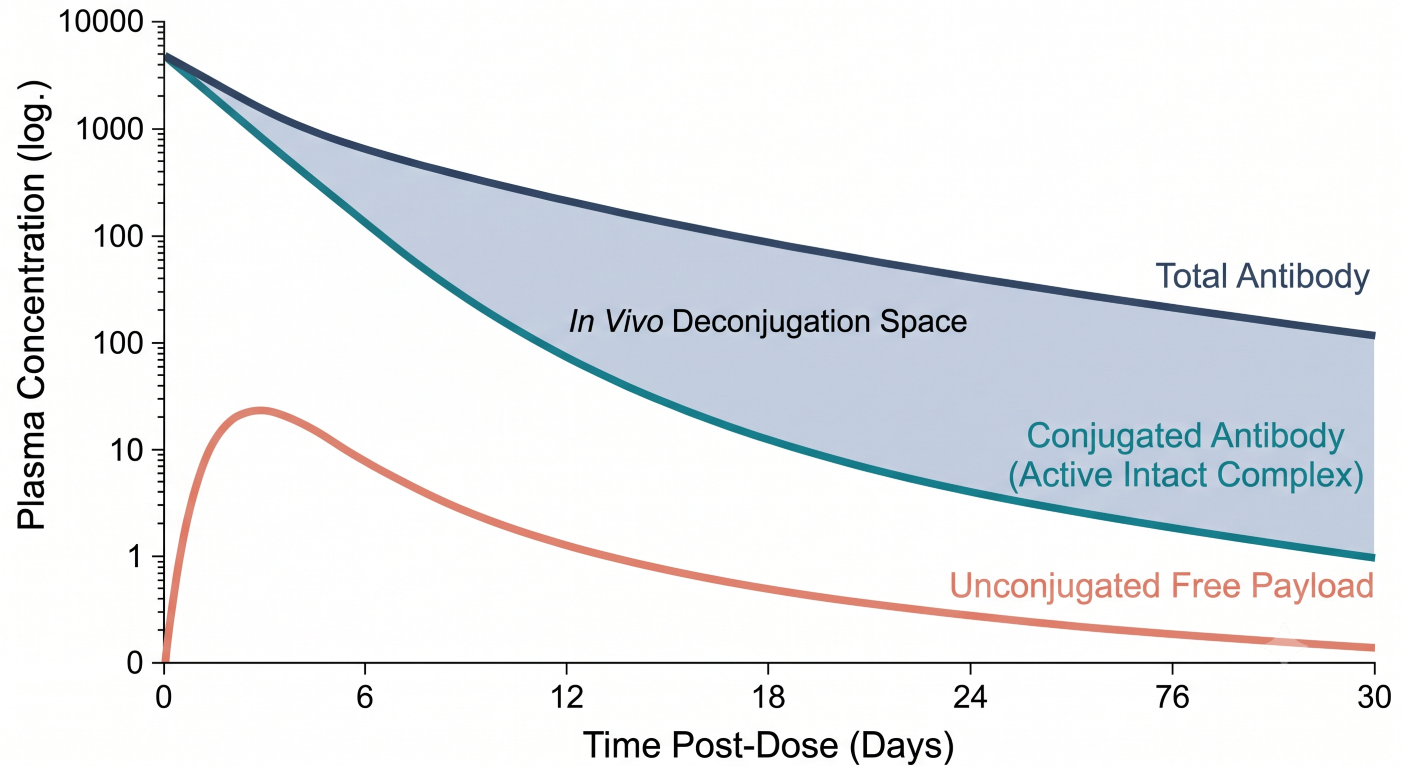
Figure 1: Pharmacokinetic Profile Showing Total Antibody, Conjugated Antibody, and Free Payload Dynamics Over Time
Characterizing the dynamic relationship between these analytes is essential to understanding the changing Drug-to-Antibody Ratio (DAR) in vivo. Over the systemic time-course, the concentration of the conjugated antibody systematically decreases at a faster rate than the total antibody pool. This divergence is driven by systemic deconjugation. The CPP must outline population pharmacokinetic frameworks designed to co-model these three structural streams simultaneously. This modeling confirms whether systemic safety signals track with the exposure metrics (AUC, Cmax) of the active complex or the accumulation kinetics of the free payload.
Section 2: Deconjugation, Linker Stability, and Target Elimination
The systemic elimination of an ADC occurs through a continuous competition between intentional target-mediated clearance and unintentional systemic linker cleavage. Linker design dictates how these pathways balance.
Cleavable linkers (such as peptide bonds highly sensitive to intracellular lysosomal cathepsin B) are optimized for rapid, high-efficiency release inside target tumor environments but are susceptible to low-level systemic enzymatic cleavage. Conversely, non-cleavable linkers (such as stable thioether bonds) require complete proteolytic breakdown of the antibody backbone within the lysosome. This structural configuration significantly minimizes systemic deconjugation but requires active intracellular peptide transport systems to utilize the payload remnant.
The intended destination for therapeutic clearance is governed by Target-Mediated Drug Disposition (TMDD). Upon high-affinity binding to the tumor-associated antigen (TAA) on the malignant cell membrane, the intact ADC complex undergoes receptor-mediated endocytosis into the endosome-lysosome compartment. At sub-therapeutic doses, this saturable pathway dominates total clearance. At therapeutic doses, the target receptors become saturated, and non-specific fluid-phase pinocytosis via vascular endothelial pathways becomes the rate-limiting elimination step for the intact antibody shell. The CPP must utilize modeling to quantify the fraction of payload delivered to target sites versus the fraction released prematurely into systemic circulation, as the latter directly correlates with classic dose-limiting clinical toxicities (such as neutropenia, thrombocytopenia, and alopecia).
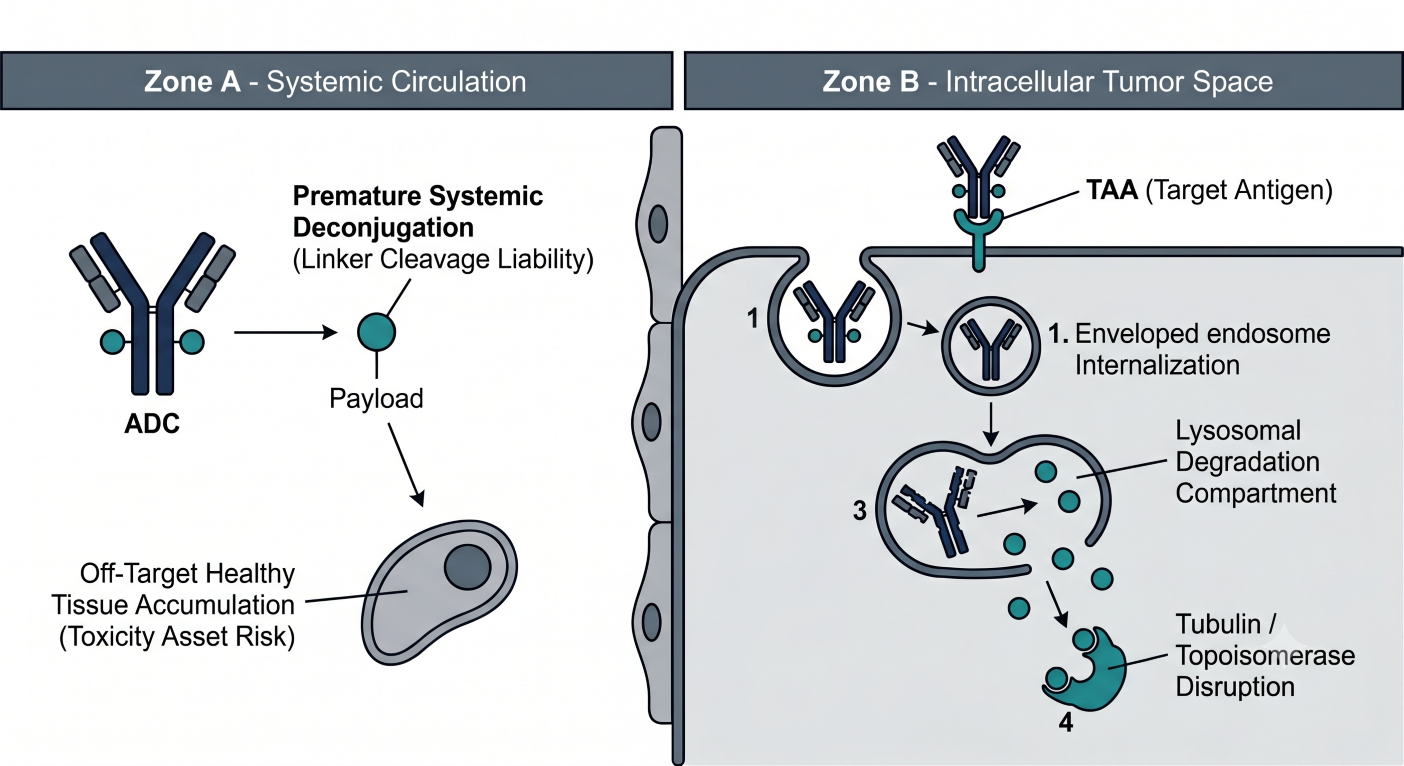
Figure 2: Cellular Processing Diagram Contrasting Target Receptor Binding and Endocytosis Against Premature Circular Payload Shedding
Section 3: Payload-Driven Extrinsic Risks and Drug-Drug Interaction Modeling
While macromolecular immunogenicity liabilities (ADA tracking) must be integrated into the portfolio, the predominant extrinsic liability in an ADC program is driven by the small molecule payload. Although the absolute circulating concentrations of the free payload are minute, frequently confined to the low picomolar range, these cytotoxic agents are exceptionally potent molecules that carry disproportionately high Drug-Drug Interaction (DDI) liabilities.
Common payloads, including monomethyl auristatin E (MMAE) and various camptothecin or exatecan derivatives (DXd), serve as high-affinity substrates and potent direct or time-dependent inhibitors of Cytochrome P450 enzymes (predominantly CYP3A4) and key efflux transporters such as P-glycoprotein (P-gp / ABCB1). Consequently, the free payload behaves as a small molecule perpetrator risk. If an ADC patient is co-administered a strong CYP3A4 inhibitor, the metabolic clearance of the free payload can be markedly suppressed. This causes an artificial elevation in the systemic exposure profile of the free toxin, leading to sudden off-target bone marrow suppression.
To navigate this without executing extensive, costly clinical drug-interaction trials, the CPP must deploy Physiologically Based Pharmacokinetic (PBPK) models parameterized with robust in vitro inhibition constants, inactivation parameters, and transporter Michaelis constants. This computational approach treats the free payload as a standalone small molecule asset within a virtual population, simulating complex clinical DDI scenarios to secure early, precise regulatory labeling and dosing guidelines.
Section 4: The Bystander Effect and Spatial Pharmacodynamics
A core objective of modern ADC engineering is the utilization of the “bystander effect” to address intratumoral heterogeneity. Solid tumor masses are rarely uniform; they consist of an intertwined matrix of antigen-positive and antigen-negative malignant cells.
When an intact ADC binds to an antigen-positive cell and undergoes lysosomal processing, the payload is liberated. If the payload molecule is structurally uncharged and highly lipophilic (such as DXd or MMAE), it can passively diffuse across the plasma membrane of the dying target cell into the surrounding extracellular tumor space. This active free payload then enters neighboring cells via passive transport, destroying adjacent antigen-negative tumor cells.
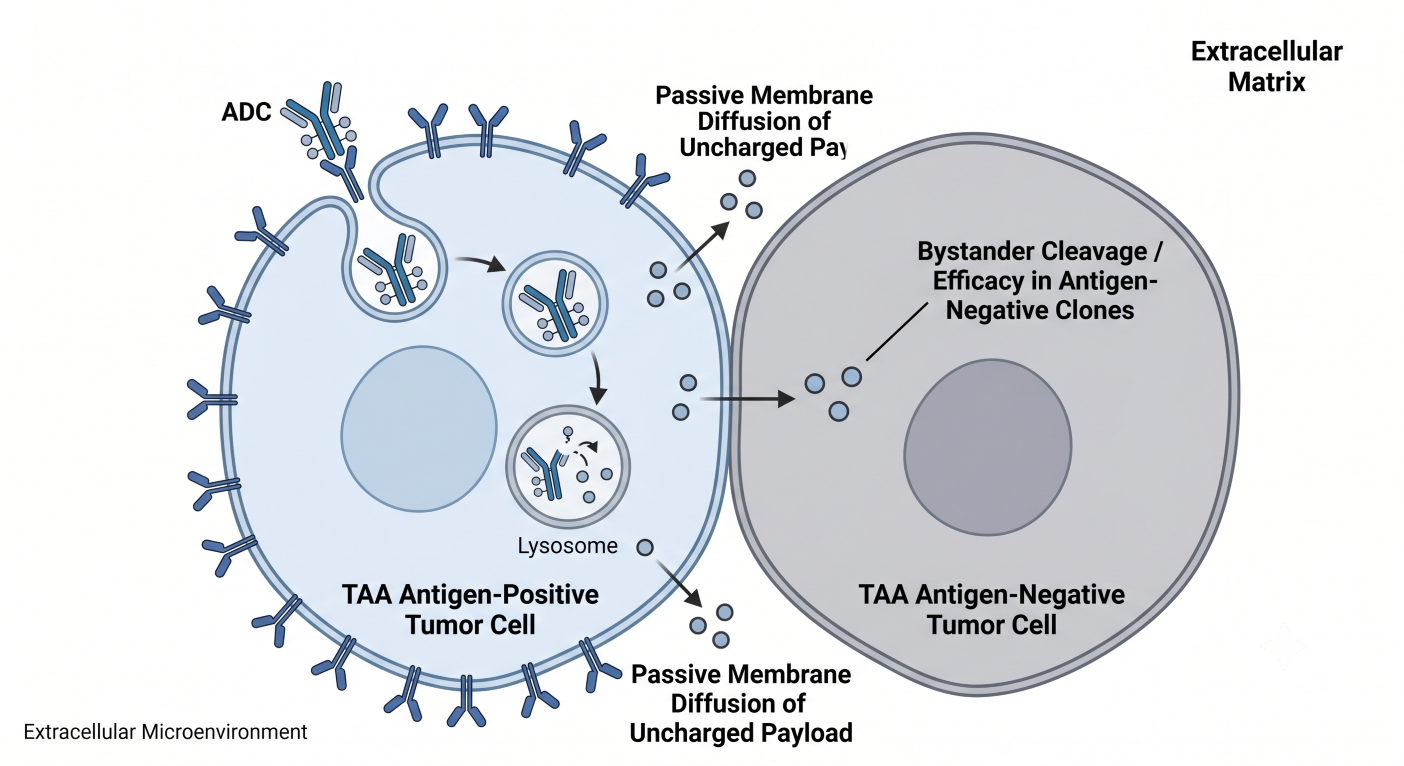
Figure 3: Spatial Synapse Rendering Depicting Lateral Membrane Diffusion of Cleaved Payload into Neighboring Antigen-Negative Clones
The CPP must mathematically balance this spatial distribution. If a payload is excessively lipophilic or if the linker chemistry displays inadequate systemic stability, the bystander effect can spill over into healthy adjacent structures, provoking localized toxicities such as interstitial lung disease (ILD) or corneal epithelial changes. Pharmacometricians must deploy spatial Quantitative Systems Pharmacology (QSP) models that combine target antigen density, internalization rates, tissue vascularity, and cellular shedding profiles to optimize the drug’s therapeutic index.
Section 5: The ADC MIDD Engine under Finalized ICH M15 Standards
The finalized ICH M15 Level 1 guidance establishes clear operational rules for quantitative evidence. Computational models utilized to support pivotal regulatory decisions must establish credibility through robust sensitivity analysis and predefined protocols. For an ADC asset, Model-Informed Drug Development (MIDD) requires an integrated multi-tiered modeling stack:
- Multi-Analyte Population Pharmacokinetics (PopPK): PopPK structures for ADCs utilize parallel differential equations to simultaneously track the elimination of the conjugated antibody, total antibody, and free payload. Crucial physiological covariates that must be captured include body surface area (BSA), baseline serum albumin (a marker of non-specific catabolic capacity), and circulating target shedding burden.
- Exposure-Response (E-R) Safety and Efficacy Integration: Driven by the FDA’s Project Optimus mandate, sponsors must move away from legacy Maximum Tolerated Dose (MTD) designs. E-R modeling for ADCs maps conjugated antibody exposure against longitudinal tumor metrics (RECIST 1.1) while simultaneously plotting systemic free payload metrics against hematological safety profiles. This dual optimization identifies the Optimal Biological Dose (OBD) early in clinical development.
- Hybrid PBPK-QSP Networks: These advanced frameworks simulate the entire physiological lifecycle of the drug. They model convective macromolecular tissue distribution across leaky tumor capillaries, local intracellular lysosomal breakdown, tubulin or topoisomerase target binding kinetics, and subsequent cellular efflux via multidrug resistance transporters (MDR1).
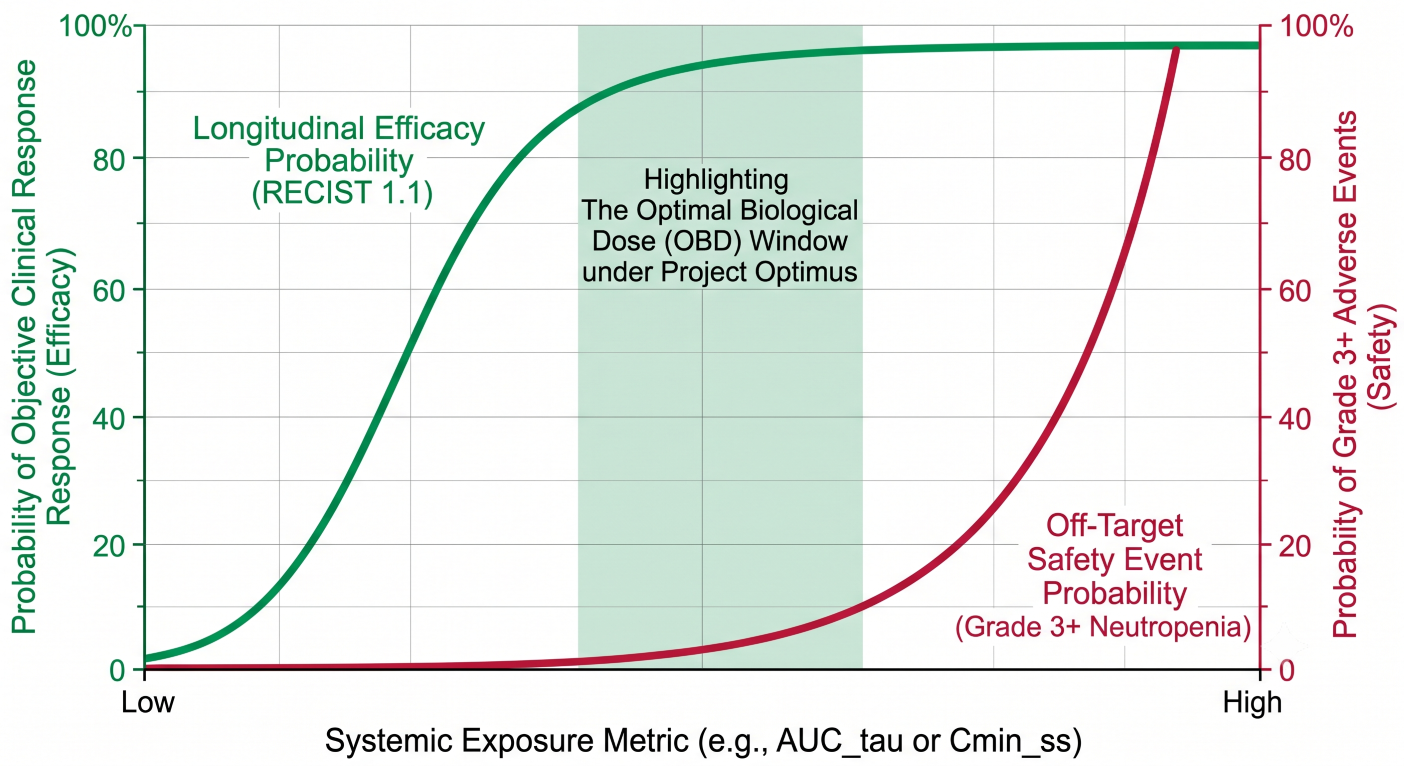
Figure 4: Dual-Axis Exposure-Response Function Curves Isolating the Optimal Biological Dose Window Under Project Optimus Guidelines
Section 6: Comparative Modality Metrics
To ensure clarity within the development portfolio, the table below contrasts the fundamental clinical pharmacology parameters of standalone monoclonal antibodies against the multi-component ADC framework.
| Pharmacokinetic Parameter | Monoclonal Antibodies (mAbs) | Antibody-Drug Conjugates (ADCs) |
|---|---|---|
| Primary Analyte Focus | Single systemic stream tracking intact, functional immunoglobulins. | Three parallel systems: Total antibody, conjugated active antibody, and unconjugated free payload. |
| Clearance Mechanisms | Nonspecific pinocytosis balanced against saturable receptor-mediated TMDD. | Standard antibody elimination pathways competed directly against systemic chemical linker deconjugation. |
| Extrinsic DDI Risk Profile | Low risk; limited to downstream cytokine-mediated CYP enzyme suppression. | High perpetrator and victim liability via released small molecule payload interactions with CYP3A4 and P-gp. |
| Primary Off-Target Toxicity | Exaggerated on-target immunomodulation or cross-reactive tissue binding. | Off-target normal tissue distribution of highly potent free toxin following systemic deconjugation. |
| Core Modeling Infrastructure | Empirical PopPK covariate modeling and minimal PBPK structures. | Multi-analyte compartment PopPK, small molecule payload PBPK, and spatial cellular QSP networks. |
Conclusion
The construction of a Clinical Pharmacology Plan for an Antibody-Drug Conjugate requires an integrated framework capable of simultaneously managing macromolecular tissue convection and small molecule systemic disposition. By implementing multi-analyte PopPK equations, modeling free payload DDI risk via PBPK networks, and using QSP to map spatial bystander mechanics, drug development teams can systematically satisfy both Project Optimus mandates and the rigorous validation standards set forth by the finalized ICH M15 guidance. This mechanistic precision replaces legacy empirical playbooks, ensuring the rapid, optimized approval of complex biological assets.
Abbreviations
ADA: Anti-Drug Antibody
ADC: Antibody-Drug Conjugate
AUC: Area Under the Curve
BSA: Body Surface Area
CPP: Clinical Pharmacology Plan
CYP: Cytochrome P450 Enzyme
DAR: Drug-to-Antibody Ratio
DDI: Drug-Drug Interaction
DXd: Exatecan Derivative Payload
E-R: Exposure-Response
ICH: International Council for Harmonisation
ILD: Interstitial Lung Disease
mAb: Monoclonal Antibody
MDR1: Multidrug Resistance Protein 1
MIDD: Model-Informed Drug Development
MTD: Maximum Tolerated Dose
OBD: Optimal Biological Dose
P-gp: P-glycoprotein Efflux Transporter
PBPK: Physiologically Based Pharmacokinetics
PopPK: Population Pharmacokinetics
QSP: Quantitative Systems Pharmacology
TAA: Tumor-Associated Antigen
TMDD: Target-Mediated Drug Disposition
Technical References
- FDA Guidance for Industry (2025). Clinical Pharmacology Considerations for Antibody-Drug Conjugates.
- FDA Guidance for Industry (2026). Optimizing the Dosage of Human Clinical Trials for Oncology Drugs Under Project Optimus.
- ICH M15 Final Guideline (2026). General Principles for Model-Informed Drug Development.
- Gibiansky, L., & Gibiansky, E. (2014). Integrated model of an antibody-drug conjugate and its payload. Journal of Pharmacokinetics and Pharmacodynamics, 41(3), 233-245.
- Shah, D. K., et al. (2012). Quantitative systems pharmacology modeling of antibody-drug conjugates. CPT: Pharmacometrics & Systems Pharmacology, 1(9), e8.