The Modality Matrix Series Portfolio
This fifth installment shifts focus to genetic medicines, addressing the unique intracellular pharmacodynamics and clearance mechanisms of synthetic nucleic acids. Managing Oligonucleotide Therapeutics requires a Clinical Pharmacology Plan (CPP) specifically calibrated to the FDA’s final June 2024 regulatory guidance.
| Article Sequence | Thematic Focus | Status |
|---|---|---|
| Article 1 | Overview of Modalities and the Clinical Pharmacology Plan | Published |
| Article 2 | Small Molecule PK and Bioavailability | Published |
| Article 3 | Large Molecule Biologics and Subcutaneous Delivery | Published |
| Article 4 | Antibody-Drug Conjugates (ADCs) and Multi-Analyte PK | Published |
| Article 5 | Oligonucleotide Therapeutics (ASOs, siRNAs, LNPs) | Current Report |
Introduction: Defining the Regulatory Scope
The FDA’s June 2024 final guidance, Clinical Pharmacology Considerations for the Development of Oligonucleotide Therapeutics, marks a critical inflection point for the biotech industry [1]. As capital continues to flow into genetic medicines, startups must recognize that transitioning these modalities into the clinic requires abandoning traditional small-molecule paradigms.
To effectively de-risk a Phase 1/2 program, developers must first explicitly define their regulatory scope. The FDA guidance specifically covers synthetically modified RNA or RNA/DNA hybrids designed to bind to a target RNA sequence via Watson-Crick base pairing (e.g., siRNAs, ASOs, and miRNAs). It explicitly excludes modalities that do not rely on base pairing for their primary mechanism of action, such as aptamers, TLR9 agonists, and CRISPR/Cas9 systems.

Figure 1: Flowchart Detailing FDA Guidance Scope for Watson-Crick Base Pairing Modalities
Section 1: The PK/PD Disconnect: Modeling for Tissue, Not Plasma
Perhaps the most significant hurdle for clinical pharmacology teams is adapting to the profound disconnect between plasma pharmacokinetics (PK) and pharmacodynamics (PD). Unlike small molecules, where plasma exposure often correlates directly with efficacy, ASOs and siRNAs clear from systemic circulation rapidly (often within hours) but accumulate heavily in target tissues. This creates a depot effect, driving a pharmacodynamic response that can last for weeks or even months [5].
This prolonged tissue-level effect must dictate the clinical dosing strategy. Relying on plasma clearance models instead of mechanistic intracellular PK/PD modeling inevitably results in incorrect, overly frequent dosing intervals. Real-world therapeutics perfectly illustrate this phenomenon:
- Inclisiran (Leqvio – siRNA): This GalNAc-conjugated siRNA targets PCSK9 mRNA in the liver. While its plasma concentrations drop to undetectable levels within 48 hours, its highly stable loading into the RNA-induced silencing complex (RISC) allows for a staggering 6-month maintenance dosing interval [5].
- Nusinersen (Spinraza – ASO): Administered intrathecally, this splice-modulating ASO clears from the CSF steadily but accumulates heavily in spinal motor neurons. Because it avoids nuclease degradation and continuously modulates pre-mRNA splicing inside the nucleus, its maintenance dose is just once every 4 months [5].
- Eplontersen (Wainua – ASO) & Pelacarsen (ASO): Both are GalNAc-conjugated ASOs. Despite rapid plasma clearance, their terminal tissue elimination half-lives support extended dosing. Eplontersen is dosed monthly [5], while Pelacarsen has shown profound target suppression with dosing intervals stretching up to 4 to 8 weeks.
- Tofersen (Qalsody – ASO): An unconjugated, RNase H-dependent ASO administered intrathecally. It remains trapped within neurons and glial cells to continuously degrade mutated SOD1 mRNA, supporting a 4-week dosing schedule [5].
Expert Note: The prolonged PD effect driven by intracellular accumulation is characteristic of ASOs (via RNase H1 cleavage or splice modulation) and siRNAs (via RISC loading). Sponsors developing LNP-mRNA therapeutics must adopt a completely different modeling paradigm, as their PD duration is dictated by the half-life of the translated protein, not the nucleic acid itself.
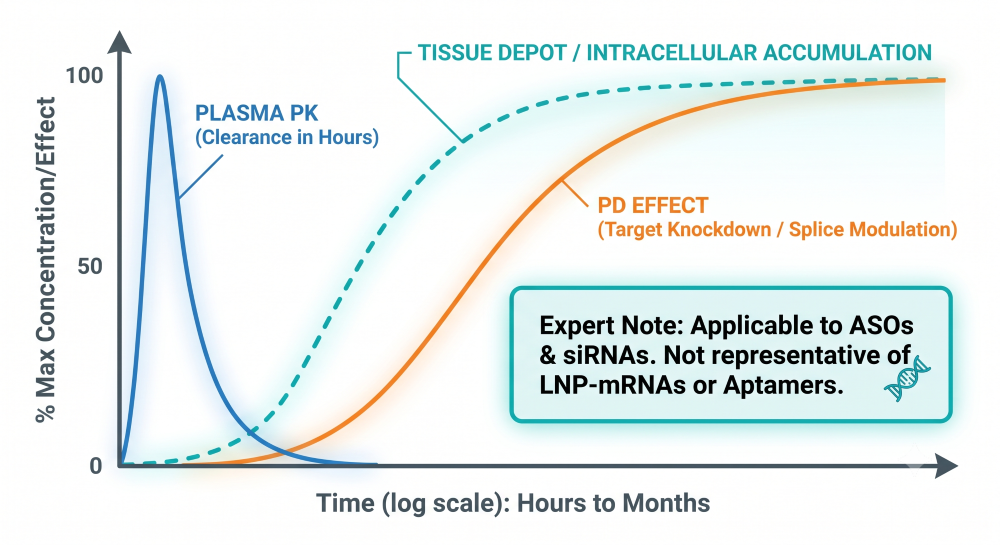
Figure 2: Fast Plasma Clearance vs. Prolonged Intracellular Tissue and PD Effects for ASOs and siRNAs
Section 2: Navigating the FDA’s Stance on QTc Prolongation
Current clinical data suggests that oligonucleotides generally have a low risk for direct hERG channel blockade. However, the FDA’s final guidance explicitly mandates a robust QT assessment plan for all oligo development programs [1]. The strategic maneuver for early-stage biotechs is to avoid the massive expense of a standalone Phase 3 Thorough QT (TQT) study by frontloading this data.
By building intensive, time-matched ECG and PK monitoring into early Single Ascending Dose (SAD) and Multiple Ascending Dose (MAD) Phase 1 cohorts, sponsors can satisfy the FDA’s requirement using a risk-based approach.
| Approach | Timing | Resource Burden | Regulatory Viability |
|---|---|---|---|
| Intensive Phase 1 ECG/PK Modeling | Phase 1 (SAD/MAD) | Low to Moderate | Highly Preferred (Risk-Based) |
| Standalone Thorough QT (TQT) Study | Phase 2 / Phase 3 | Extremely High | Required only if signal detected |
Table 1: QTc Assessment Strategies for Early-Phase Oligo Trials
Section 3: Immunogenicity Risk Assessments: The GalNAc vs. LNP Divide
Not all delivery systems are created equal, and your Clinical Pharmacology Plan must reflect the reality of your specific modality. Applying a “one-size-fits-all” immunogenicity strategy will either unnecessarily bloat your Phase 1 budget or trigger a clinical hold.
Based on extensive clinical experience, GalNAc-conjugated oligos present a largely “silent” immunogenic profile. Because GalNAc utilizes a natural sugar moiety targeting the ASGPR receptor on hepatocytes, and the oligonucleotides themselves utilize stealth chemistry (e.g., 2′-O-methyl modifications), they rarely elicit meaningful anti-drug antibody (ADA) or innate immune responses [2].
Conversely, Lipid Nanoparticles (LNPs) carry a significant immunogenic burden. The systemic administration of LNPs can trigger Toll-like Receptors (TLRs) and complement activation, driving the upregulation of pro-inflammatory cytokines like IL-6 and TNF-α [3]. Sponsors utilizing novel LNPs must adopt a rigorous, multi-tiered ADA and cytokine assessment plan.
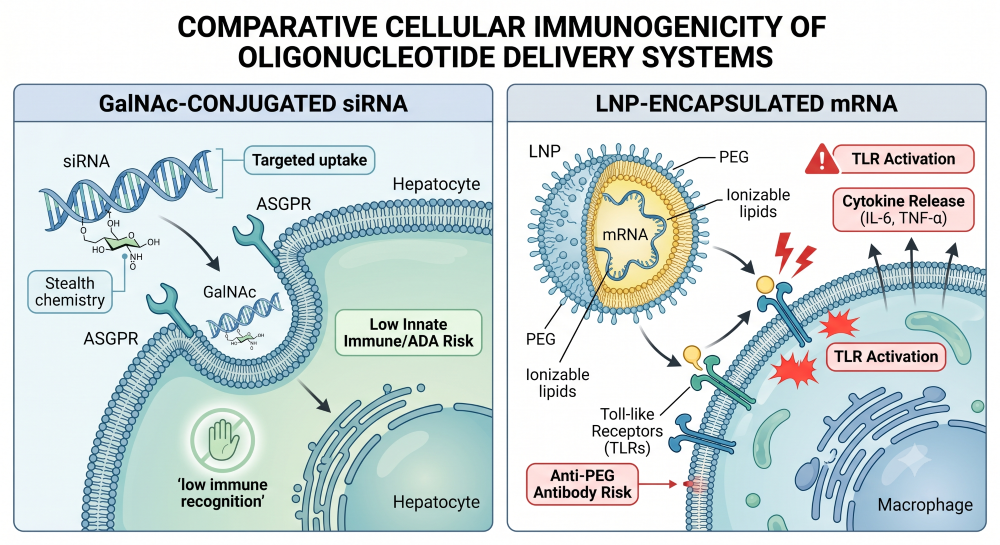
Figure 3: Visual Comparison of GalNAc ASGPR Uptake vs. LNP Immune Activation
Section 4: Rethinking Drug Interactions and Organ Impairment Strategies
Because oligonucleotides are metabolized universally by ubiquitous tissue endo- and exonucleases rather than the Cytochrome P450 (CYP) system, traditional drug-drug interaction (DDI) risks are incredibly low. Early in vitro data is usually sufficient to bypass the need for extensive clinical CYP or transporter DDI studies. However, predicting where the drug will accumulate dictates your organ impairment strategy.
Clearance pathways and clinical impairment risks are highly modality-dependent:
- LNP-mRNAs: These large macromolecular complexes escape renal filtration entirely and are primarily cleared by the reticuloendothelial system (RES) and macrophages. Consequently, specific organ impairment dose adjustments are rarely a clinical focus.
- GalNAc-siRNAs: Designed for rapid, high-affinity uptake via the ASGPR receptor, these molecules partition almost entirely into the liver where they are metabolized by intracellular nucleases. While a minor fraction is excreted renally, clinical precedents (e.g., inclisiran, givosiran) demonstrate that even severe renal impairment does not require dose adjustments, as increased plasma AUC does not alter the hepatic pharmacodynamic effect or drive toxicity.
- ASOs: Due to backbone modifications (like phosphorothioate linkages), ASOs bind heavily to plasma proteins, preventing rapid filtration. Instead, they distribute to tissues and accumulate significantly in the proximal tubule cells of the kidney. In patients with severe renal impairment, systemic clearance is reduced, which compounds tubular accumulation and nephrotoxicity risks. Rigorous renal PK assessments are non-negotiable here.
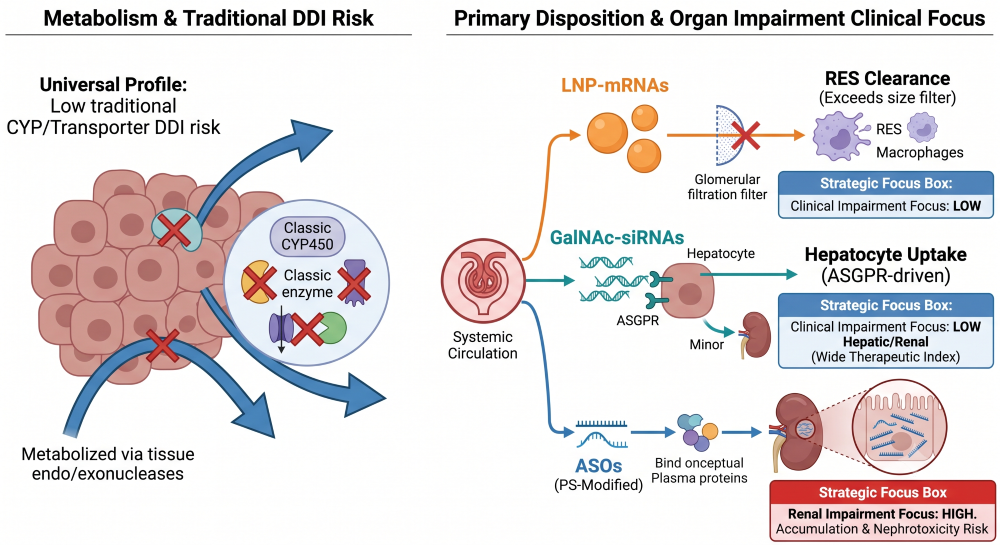
Figure 4: Low CYP DDI Risk Alongside RES, Hepatic, and Renal Clearance Paths
| Modality | Primary Clearance / Tissue Depot | Organ Impairment Dose Adjustment Focus? |
|---|---|---|
| LNP-mRNAs | RES / Macrophages | Low (Exceeds filtration threshold; RES driven) |
| GalNAc-siRNAs | Hepatocyte (ASGPR) / Intracellular Nucleases | Low (Wide TI; increased plasma AUC rarely impacts safety) |
| ASOs | Tissue uptake / Proximal Tubules | High Renal Focus (Tubular accumulation & nephrotoxicity risk) |
Table 2: Matrix of Modality-Specific Impairment Strategies
Abbreviations
ADA: Anti-Drug Antibody
ASGPR: Asialoglycoprotein Receptor
ASO: Antisense Oligonucleotide
CPP: Clinical Pharmacology Plan
CSF: Cerebrospinal Fluid
CYP: Cytochrome P450
DDI: Drug-Drug Interaction
ECG: Electrocardiogram
FDA: Food and Drug Administration
GalNAc: N-acetylgalactosamine
LNP: Lipid Nanoparticle
MAD: Multiple Ascending Dose
miRNA: microRNA
mRNA: Messenger RNA
PD: Pharmacodynamics
PK: Pharmacokinetics
RES: Reticuloendothelial System
RISC: RNA-Induced Silencing Complex
SAD: Single Ascending Dose
siRNA: Small Interfering RNA
TLR: Toll-Like Receptor
TQT: Thorough QT
Technical References
- U.S. Food and Drug Administration (FDA). (2024, June). Clinical Pharmacology Considerations for the Development of Oligonucleotide Therapeutics. Guidance for Industry.
- Li, J., & Foged, C. (2024). Evaluating the breadth of nucleic acid-based payloads delivered in lipid nanoparticles to establish fundamental differences in development. Expert Opinion on Drug Delivery, 21, 1441–1461.
- Ranjbar, S., Zhong, X., Manautou, J., & Lu, X. (2023). A holistic analysis of the intrinsic and delivery-mediated toxicity of siRNA therapeutics. Advanced Drug Delivery Reviews, 201, 115052.
- Shen, L., et al. (2023). Nephrotoxicity of marketed antisense oligonucleotide drugs: A comprehensive review of clearance mechanisms and clinical implications. Toxicology.
- U.S. Food and Drug Administration. Prescribing Information for Approved Oligonucleotide Therapeutics: Leqvio (inclisiran), Spinraza (nusinersen), Wainua (eplontersen), and Qalsody (tofersen).