
Introduction
The term “me-too” drug has historically been used pejoratively to describe follow-on compounds perceived as redundant or commercially motivated imitations of first-in-class agents. Yet this view overlooks a critical reality of modern drug development: once a pathway or target is clinically validated, subsequent entrants can capitalize on existing biological, clinical, and regulatory knowledge to accelerate development, reduce risk, and refine therapeutic profiles [1]. These drugs, often better termed next-in-class or me-better, can meaningfully expand patient options and, in many cases, achieve improvements over their pioneering predecessors.
This article examines the value of me-too drug development from a drug development strategy perspective. Rather than focusing solely on market competition, we highlight how companies leverage validated pathways to streamline development, enhance safety and efficacy profiles, and expand indications. Examples span small molecules, biologics, antibody–drug conjugates (ADCs), and RNA-based therapies, illustrating how thoughtful follow-on strategies can yield substantial patient and commercial impact.
Advantages of Targeting Validated Pathways
Reduced Preclinical and Translational Risk
First-in-class drugs often establish proof-of-biology for a target, confirming that modulation yields clinical benefit [1]. Follow-on agents can bypass much of this uncertainty, focusing preclinical work on differentiation (improving potency, selectivity, or delivery) rather than revalidating the pathway. This reduces attrition risk and optimizes resource allocation.
For example, after the first TNF inhibitor infliximab transformed autoimmune disease treatment, subsequent agents like adalimumab and etanercept leveraged extensive TNF biology to prioritize innovations in format and immunogenicity [2].
Streamlined Clinical Development
Validated targets also enable accelerated clinical programs: biomarkers are defined, endpoints are standardized, and anticipated safety profiles are known. This facilitates earlier go/no-go decisions, adaptive designs, and even abbreviated development timelines. Regulatory agencies frequently extrapolate class experience to support accelerated approvals, particularly in oncology and rare diseases.
Regulatory Precedent and CMC Learnings
Follow-on drugs benefit from regulatory precedents set by the first entrant. Prior approvals guide reviewers’ expectations for safety monitoring and study designs, while CMC learnings inform formulation refinements (e.g., stability, immunogenicity reduction) [1]. This synergy can significantly shorten the path to market.
Case Studies in Leveraging Validated Pathways
PD-1/PD-L1 Checkpoint Inhibitors: Building on Immuno-Oncology Success
The immune checkpoint inhibitor revolution began with CTLA-4 and PD-1 blockade. After pembrolizumab and nivolumab validated PD-1 as a therapeutic target, multiple PD-1/PD-L1 antibodies followed (atezolizumab, durvalumab, cemiplimab, avelumab) [3]. These me-too programs leveraged established biomarkers (PD-L1 IHC), toxicity management frameworks (immune-related adverse events), and clinical endpoints (overall survival in specific tumors) to expedite development. While later entrants often lacked major efficacy advantages, they expanded access to immunotherapy across tumor types and facilitated indication-specific differentiation.
TNF Inhibitors: Iterative Improvements in Autoimmune Disease
The chimeric monoclonal antibody infliximab was the first TNF inhibitor approved for rheumatoid arthritis and Crohn’s disease. Building on its success, fully human antibodies (adalimumab, golimumab) and receptor-Fc fusion proteins (etanercept) entered the market [2]. These follow-ons benefitted from extensive TNF biology and safety data, focusing innovation on self-injectable formulations and reduced immunogenicity. The result was a robust class with multiple therapeutic options, and adalimumab ultimately became the world’s top-selling drug for over a decade.
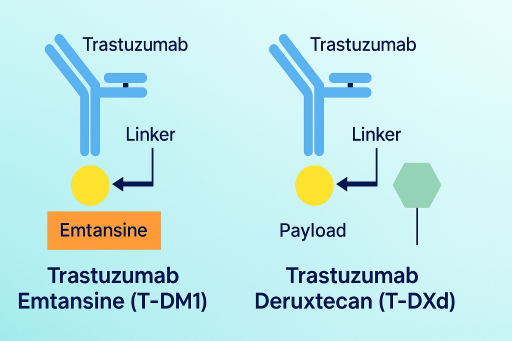
HER2-Targeted Antibody–Drug Conjugates: From Trastuzumab to T-DXd
Trastuzumab established HER2 as a critical breast cancer target. The follow-on ADC trastuzumab emtansine (T-DM1) combined trastuzumab with a cytotoxic payload, offering targeted chemotherapy and improving outcomes in second-line settings [4]. The next iteration, trastuzumab deruxtecan (T-DXd), introduced a more potent payload and cleavable linker, resulting in a dramatic efficacy improvement: in DESTINY-Breast03, 12-month progression-free survival was 75.8% with T-DXd vs 34.1% with T-DM1 (HR 0.28; P<0.001) [4]. Here, me-too development not only leveraged existing HER2 biology but redefined the class standard, illustrating how iterative innovation can transform therapeutic impact.
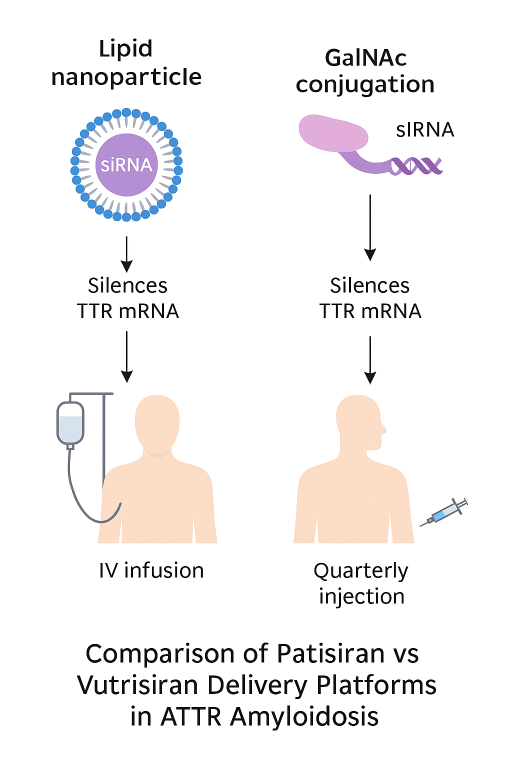
RNA Therapeutics for TTR Amyloidosis: Vutrisiran’s Leap Beyond Patisiran
Transthyretin (TTR) amyloidosis, encompassing hereditary (hATTR) and wild-type (ATTRwt) forms, is a progressive and often fatal disease driven by TTR protein deposition in nerves and heart tissue. The first RNAi therapeutic, patisiran (Onpattro), validated TTR knockdown as a transformative approach when approved in 2018 for hATTR polyneuropathy [5].
Leveraging Pathway Validation
Patisiran’s success catalyzed rapid development of next-generation agents, including inotersen (antisense oligonucleotide) and vutrisiran (Amvuttra), an siRNA conjugated to GalNAc for subcutaneous quarterly dosing [6]. By leveraging established knowledge such as validated biomarkers (mNIS+7, NT-proBNP), safety expectations, and regulatory precedent, vutrisiran’s program accelerated without needing to re-establish target validity.
Broader Indications and Regulatory Milestones
In March 2025, the U.S. FDA approved vutrisiran for both hereditary and wild-type ATTR cardiomyopathy (ATTR-CM), expanding beyond neuropathy to the broader cardiac phenotype [7]. The European Commission followed in June 2025. Approval was based on HELIOS-B, which demonstrated:
- Approximately 28–36% reduction in all-cause mortality vs placebo
- Significant reduction in heart-failure hospitalizations and urgent visits
- Improved functional outcomes (6-minute walk test, quality-of-life scores) across patient populations
Development and Commercial Advantages
Vutrisiran’s advantages over patisiran are multifaceted:
- Route and frequency: Subcutaneous dosing every 3 months vs patisiran’s intravenous infusions every 3 weeks (with premedication)
- Broader indication: Approved for both neuropathy and cardiomyopathy, whereas patisiran remains neuropathy-only
- Healthcare efficiency: Eliminates infusion center requirements, lowering burden for patients and providers
- Market reach: ATTR-CM is more prevalent than hATTR-PN, vastly expanding eligible patients
Balancing Benefits and Pitfalls
While validated pathways offer compelling efficiencies, me-too strategies carry inherent challenges:
- Minimal differentiation risk: Without clear advantages, follow-ons may saturate markets without improving care
- Safety complacency: Class assumptions can obscure compound-specific toxicities (e.g., rofecoxib vs celecoxib in COX-2 inhibitors)
- Regulatory scrutiny: Agencies now expect differentiation; the FDA’s rejection of sintilimab (a PD-1 inhibitor with non-U.S. trial data) exemplifies higher evidentiary bars in crowded classes
Regulatory and Strategic Perspectives
Regulators increasingly distinguish me-better from purely duplicative me-too drugs [1]. FDA and EMA leverage class precedents to expedite review but expect new entrants to add clinical or practical value. Developers strategically exploit real-world data, validated biomarkers, and surrogate endpoints to design smaller, faster trials, while pursuing differentiation through formulation improvements or indication expansion (as with vutrisiran).
Conclusion
Me-too drug development, when viewed through a scientific and strategic lens, is less about imitation than optimization and acceleration. By leveraging validated targets, companies reduce risk, shorten timelines, and often deliver superior therapies [1]. The evolution of vutrisiran from patisiran exemplifies this trajectory: starting with pathway validation, advancing through formulation innovation, and culminating in broader indications and clinical convenience.
In today’s drug development landscape, especially in rare diseases and oncology, thoughtful me-too strategies complement first-in-class breakthroughs. The key is ensuring that follow-on drugs provide genuine value to patients and healthcare systems, transforming what was once seen as “copycat” into a cornerstone of therapeutic progress.
References
- Aronson JK, Green AR. Me-too pharmaceutical products: history, definitions, examples, and relevance. Br J Clin Pharmacol. 2020;86(11):2114–2122.
- Feldmann M, Maini RN. Anti-TNF therapy, from rationale to standard of care: what lessons has it taught us? Immunol Rev. 2008;223:9–23.
- Olivier T et al. Oncology “me-too” drugs versus original drugs in randomized trials. JAMA Intern Med. 2025;185(2):201–210.
- Cortes J et al. Trastuzumab deruxtecan vs trastuzumab emtansine for HER2-positive breast cancer. N Engl J Med. 2022;387:1143–1154.
- Adams D et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21.
- Alnylam Pharmaceuticals. HELIOS-A Study: Vutrisiran in hATTR amyloidosis polyneuropathy. N Engl J Med. 2022;387:1037–1048.
- Alnylam Pharmaceuticals. HELIOS-B Study: Vutrisiran in ATTR cardiomyopathy. Press release, FDA approval March 2025, EC approval June 2025. Available at: https://investors.alnylam.com.
- FDA Drug Approval Package: Amvuttra (vutrisiran) expanded indication, March 2025.