Dossier Deep Dive · Part 3
Executive Snapshot
- Why 2.7.2 matters: This is the concise, integrated clinical pharmacology narrative that underpins dose, regimen, and labeling—read alongside the Clinical Overview (2.5) and substantiated by Module 5.
- Start at IND: Draft a light “2.7.2” scaffold (exposure metrics, covariates, DDI/food-effect plan, modeling shells) and harden iteratively through EoP2.
- What reviewers expect: Clear exposure–response reasoning, population PK learnings, DDI/food-effect conclusions, and crisp linkage to the proposed label.
- Outcome: Fewer inconsistencies with Modules 2.4/2.5 and more focused questions on benefit–risk rather than re-analysis.
What 2.7.2 Is (and Isn’t)
2.7.2 = the Summary of Clinical Pharmacology within Module 2. It distills ADME, exposure metrics, dose proportionality, E–R for efficacy/safety, intrinsic/extrinsic factors, DDI, and key modeling (popPK, PBPK) into a coherent justification for the proposed dose and regimen that is tightly linked to 2.5 and substantiated in Module 5.
It’s not a data dump. It’s the argument for dose and use.
IND → EoP2 → NDA: Your 2.7.2 Roadmap
IND (plan and scaffold)
- Define exposure metrics (AUC, Cmax, Ctrough) and candidate covariates (body size, formulation, organ function, genotype, etc.).
- Outline DDI strategy (CYP/transporters), PBPK use-cases, and clinical studies to confirm/negate risks.
- Draft food-effect plan (timing/design; fed vs fasted implications).
- Create figure/table shells for E–R and popPK; decide graphics conventions now (units, scales, legends).
- Draft the 2.7.2-lite narrative: section headings and cross-reference notes to future Module 5 reports.
End of Phase 2 (populate and converge)
- Populate with Phase 1/2 popPK, E–R for efficacy/safety, intrinsic factors (renal/hepatic/PGx) updates, DDI and food-effect results.
- Converge on dose/regimen and pre-specify pivotal E–R analyses for confirmatory trials.
- Align with Module 2.5 so dose rationale and benefit–risk framing are consistent.
NDA/MAA (finalize and cross-link)
- Finalize label-ready dose/regimen rationale with integrated E–R and finalized popPK; include PBPK where appropriate.
- Cross-link statements to 2.5 and the relevant Module 5 study reports.
- Keep draft labeling language (Sections 2 and 12) traceable to 2.7.2 evidence.
Best Practices (That Pay Off at NDA)
- Write 2.7.2 as the dose spine. Lock exposure metrics early and keep them consistent across analyses/figures.
- Prospectively plan E–R. Put figure shells in an Appendix at IND so updates are data drops, not rewrites.
- Integrate with CMC changes. If process changes could alter exposure, co-author QOS (2.3) and 2.7.2 text to show how you’ll monitor and interpret impact.
- Use PBPK and popPK strategically. PBPK for DDI/special-population what-ifs; popPK to quantify variability and covariates.
- Make labeling your north star. Draft the Clinical Pharmacology (12) and Dosage & Administration (2) language you’re aiming for and back-chain analyses to support it.
- Keep terminology synchronized. Units, exposure metrics, and abbreviations must match across 2.7.2, 2.5, and Module 5.
- Maintain a lightweight change log—what changed, why, and impact on dose/regimen.
Common Pitfalls & How to Avoid Them
- Data recitation without conclusions → Always state what the result means for dosing, monitoring, or labeling.
- Inconsistent terminology/units → Maintain a central glossary and enforce it at each update.
- Fragmented DDI story → Tie in vitro → PBPK → clinical DDI results into one narrative with explicit label impact.
- Late E–R modeling → Plan datasets/graphics at IND; don’t wait for pivotal lock.
- Poor linkage to CMC comparability → If exposure could shift, show how you’ll detect and interpret it and reference QOS (2.3).
Worked Example (Fictional) — How to Write 2.7.2 for a Real Program
Molecule/Target/Indication: an example oral kinase inhibitor
Nonclinical rationale: In vitro and xenograft models show potent target inhibition with tumor growth suppression at exposures achievable in humans; hepatic and GI findings are target-organ risks at high exposures.
Clinical program: Phase 1/2 (40–200 mg QD) with expansion at 120 mg QD; pivotal single-arm confirmatory at 120 mg QD (ORR/DOR); supportive FE, DDI (CYP3A4 inhibitor/inducer), renal/hepatic impairment, and concentration–QTc modeling.
2.7.2.1 Overview and Key Findings
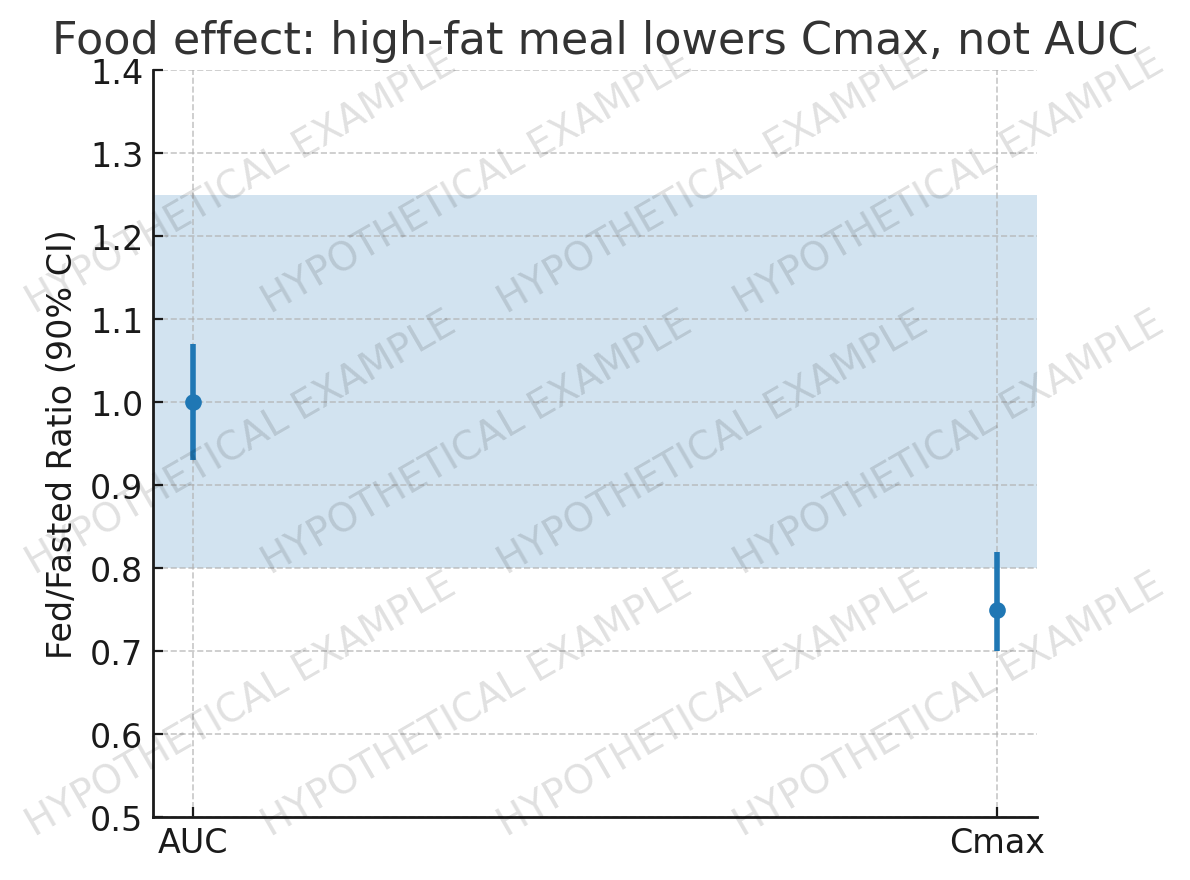
Recommended dose: 120 mg QD based on integrated E–R for efficacy (ORR) and safety (ALT/AST, diarrhea) and popPK indicating predictable exposure with moderate variability. Exposure is ~dose-proportional from 40–160 mg; t½ ≈ 14 h supports QD dosing. A high-fat meal reduces Cmax ~25% with no meaningful AUC change; dosing with or without food is acceptable. The drug is primarily metabolized by CYP3A4; use with strong CYP3A4 inhibitors/inducers requires dose modification or avoidance. Moderate hepatic impairment increases AUC ~2-fold → a lower starting dose is recommended. The totality of data supports 120 mg QD as the label dose.
2.7.2.2 Pharmacokinetics in Humans
Absorption: median Tmax ~2 h; accumulation consistent with t½.
Distribution: extensive (PPB ~95%).
Metabolism: predominantly CYP3A4; minor UGT contribution.
Excretion: <5% unchanged in urine; fecal metabolites predominate.
Dose proportionality & time dependence: ~proportional AUC/Cmax 40–160 mg QD; no time-dependent PK at steady state.
Food effect: high-fat meal ↓Cmax ~25% (AUC unchanged) → no meal restrictions proposed.
2.7.2.3 Population Pharmacokinetics
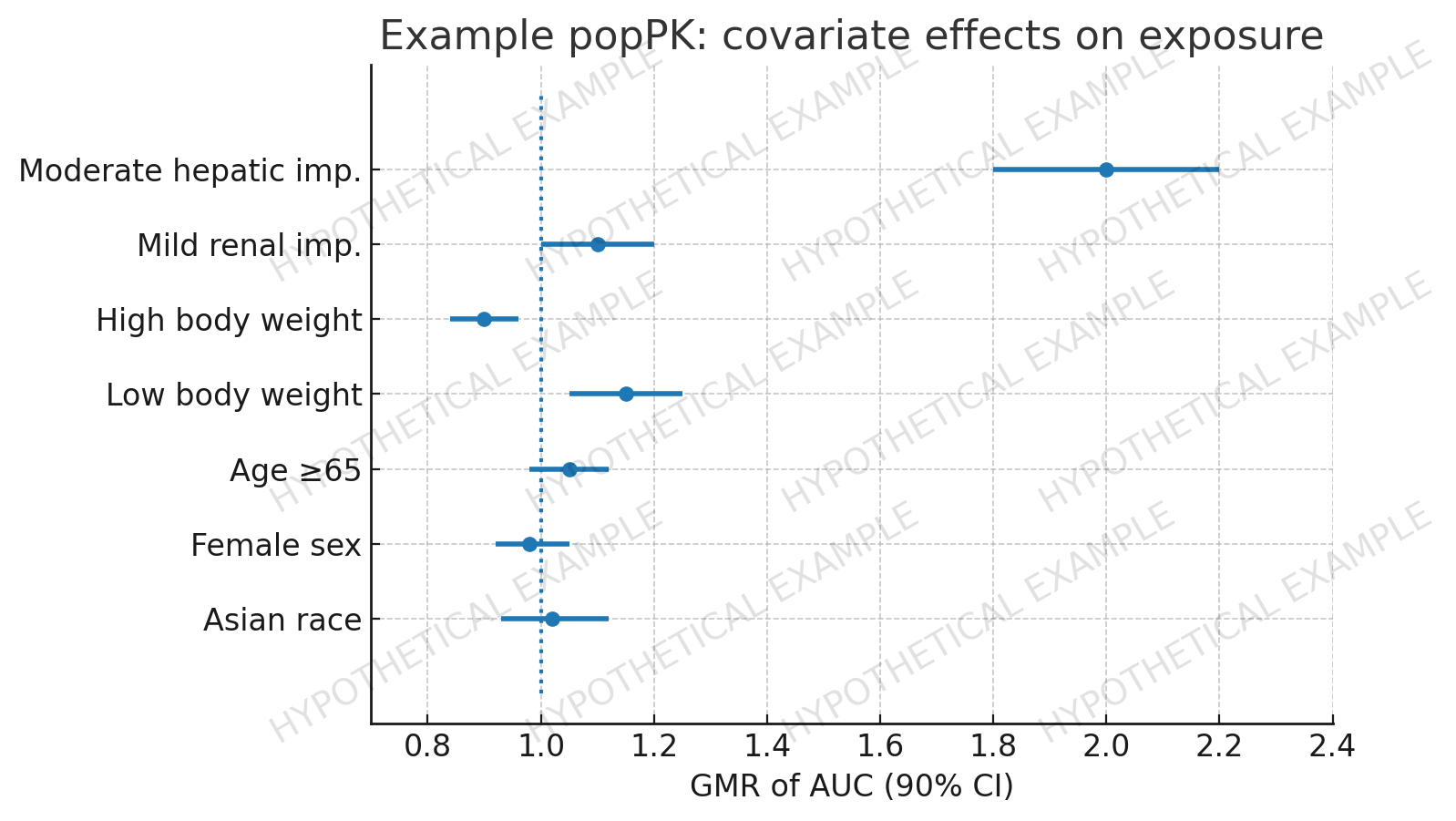
One-compartment model with first-order processes; BSV on CL/F ~35%. Covariates: body weight (small negative effect on exposure), moderate hepatic impairment (AUC ↑~2×). Age, sex, race, mild renal impairment not clinically meaningful.
2.7.2.4 Exposure–Response (Efficacy & Safety)
Efficacy: Logistic E–R shows ORR increases with AUC to a plateau; EC90 aligns with median AUC at 120 mg QD.
Safety: Higher AUC increases probability of grade ≥3 ALT/AST and diarrhea; 160 mg crosses the safety inflection.
Integration: 120 mg QD balances efficacy plateau attainment with acceptable safety risk, consistent with 2.5 benefit–risk.

2.7.2.5 Intrinsic & Extrinsic Factors
Intrinsic
• Renal impairment: mild—no meaningful effect; moderate—no starting-dose change.
• Hepatic impairment: moderate AUC ↑~2× → reduced starting dose; severe—not studied (not recommended).
• Age/Sex/Race: no clinically relevant effects after covariate adjustment.
Extrinsic
• DDI (CYP3A4): strong inhibitors ↑AUC ~2–3× (avoid or reduce dose); strong inducers ↓AUC substantially (avoid).
• Transporters: likely P-gp substrate; no meaningful clinical impact at label dose.
• Food: no clinically meaningful AUC change; with or without food.

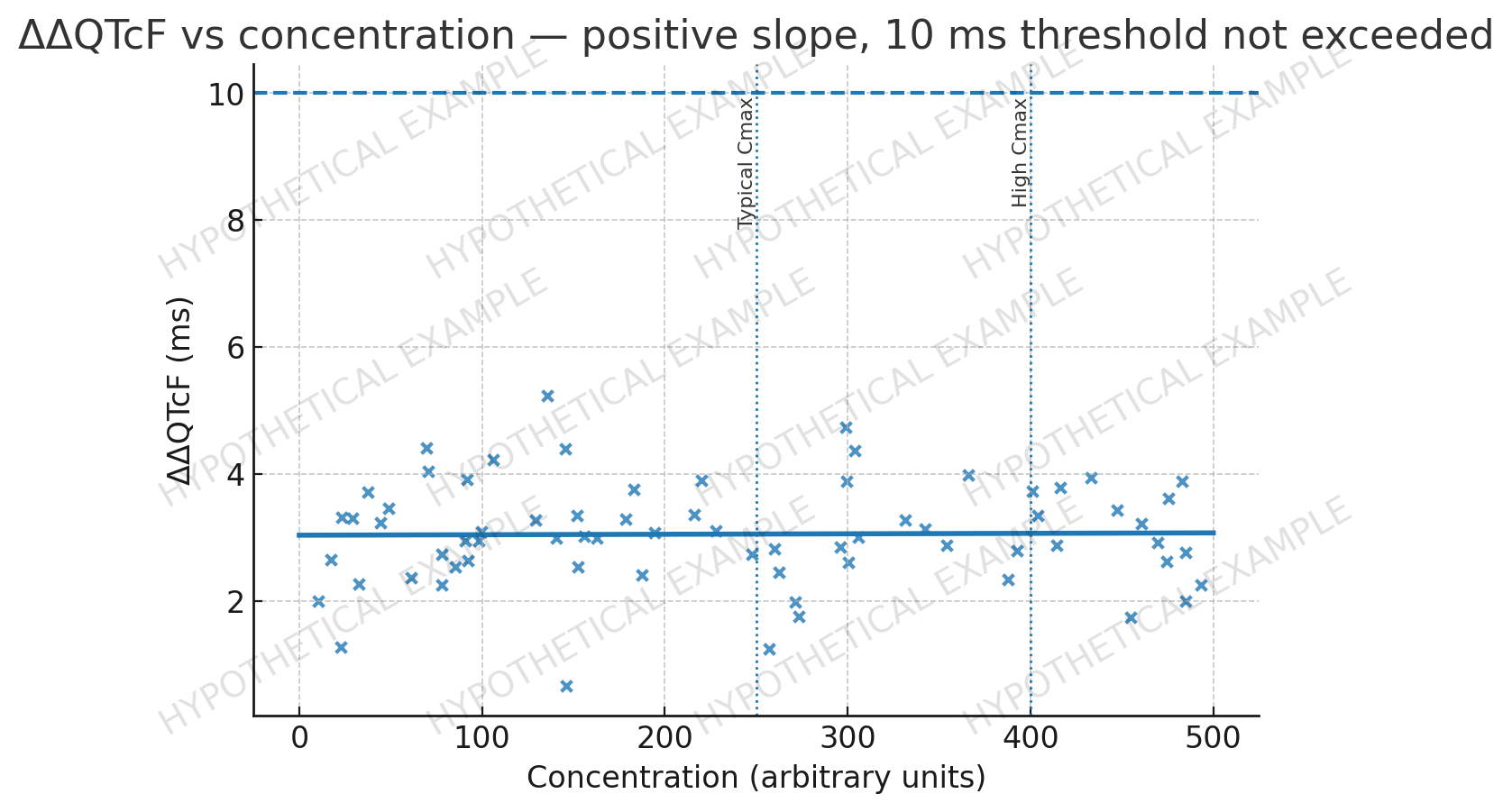
2.7.2.6 QTc and Cardiac Safety
Concentration–QTc modeling indicates a small, non-clinically meaningful slope; predicted ΔΔQTcF at typical and high exposures remains below thresholds of regulatory concern.
2.7.2.7 Special Populations and Pediatrics
No pediatric studies conducted. In ≥65 y, exposure similar to overall population after body-size adjustment; no starting-dose change based solely on age.
2.7.2.8 Dose and Regimen Selection (Label-Ready Rationale)
Recommended dose: 120 mg QD.
- Achieves exposures above EC90 for most patients.
- Remains below exposures with steep grade ≥3 ALT/AST and diarrhea risk.
- Compatible with real-world co-medications with CYP3A4 management.
- Labeling: reduced starting dose in moderate hepatic impairment; severe hepatic impairment not recommended.
Draft labeling statements (Sections 2 & 12)
• “Recommended dosage is 120 mg once daily, with or without food.”
• “Avoid strong CYP3A4 inducers; reduce dose or monitor if strong CYP3A4 inhibitors cannot be avoided.”
• “Reduce starting dose in moderate hepatic impairment; monitor liver function.”
• “No dosage adjustment for mild renal impairment, age, sex, or race.”
IND-Stage Variant (“2.7.2-lite”)
- Define exposure metrics/covariates; create E–R and popPK shells.
- Outline CYP3A4 DDI and food-effect plans; state PBPK/popPK roles.
- Pre-specify the dose-selection algorithm (e.g., lowest dose achieving EC90 in ≥80% while below safety inflection).
Practical IND Checklist for 2.7.2
- Define exposure metrics, covariates, analysis populations; create E–R and popPK shells.
- Lay out DDI plan (CYP/transporters), including PBPK-supported vs clinical studies.
- Plan food-effect timing/design (fed vs fasted statements are ultimately labeling).
- Draft target labeling (Sections 2 & 12) so analyses ladder up to actionable dosing language.
- Set an update cadence (e.g., within 30 days of each major report).
Sources
- ICH M4E(R2): CTD — Efficacy (Module 2, Clinical Overview).
- FDA. Exposure–Response Relationships — Study Design, Data Analysis, and Regulatory Applications (Guidance).
- FDA. Clinical Drug Interaction Studies — CYP-/Transporter-Mediated (Guidance).
- FDA. Population Pharmacokinetics (Final Guidance, 2022).
- FDA. Assessing the Effects of Food on Drugs (Guidance).
- FDA. Physiologically Based Pharmacokinetic Analyses — Format & Content (Guidance).
- FDA. Clinical Pharmacology Labeling — Content & Format (Guidance).
- FDA. Multidiscipline Review — Sotorasib (NDA 214665).