The CMC Story Without the Rework
1. Introduction
In our previous installment, we noted: “Next: QOS (Module 2.3) – CMC story without the rework (coming soon).”
This is that piece.
The Quality Overall Summary (QOS; Module 2.3 of the CTD) is often seen as a condensed version of Module 3 (Quality/CMC). In practice, it plays a more strategic role. The QOS is the narrative bridge between chemistry, manufacturing, and controls (CMC) and clinical relevance.
A good QOS avoids unnecessary repetition of Module 3 and instead weaves in clinical pharmacology and biopharmaceutics insights. This shows regulators how product attributes influence patient exposure, variability, and outcomes.
2. What is the QOS?
Purpose
- Summarize Module 3 in a concise, reviewer-friendly format
- Highlight quality attributes and their clinical implications
Structure
- 2.3.S – Drug Substance
- 2.3.P – Drug Product
- 2.3.A – Appendices
- 2.3.R – Regional Information
3. Role of Biopharmaceutics in the QOS
- Solubility and Dissolution: BCS classification informs formulation choices. Dissolution specifications must link to bioavailability and bioequivalence outcomes.
- Particle Size and Polymorphs: Solid-state properties often explain absorption variability or food effect.
- Excipients: Some excipients influence absorption or drug interactions.
In a strong QOS, these data are presented in the context of clinical performance.
4. Clinical Pharmacology Inputs into the QOS
- Dose and Strength Justification: Exposure-Response analyses support the dose range and choice of strengths.
- Variability and Specifications: PopPK data show when variability justifies tighter control of dissolution or release.
- Formulation Bridging: Bioequivalence and PopPK confirm the link between early and final formulations.
- Modeling and Simulation: PBPK and Exposure-Response analyses support waivers and demonstrate clinical relevance of specifications.
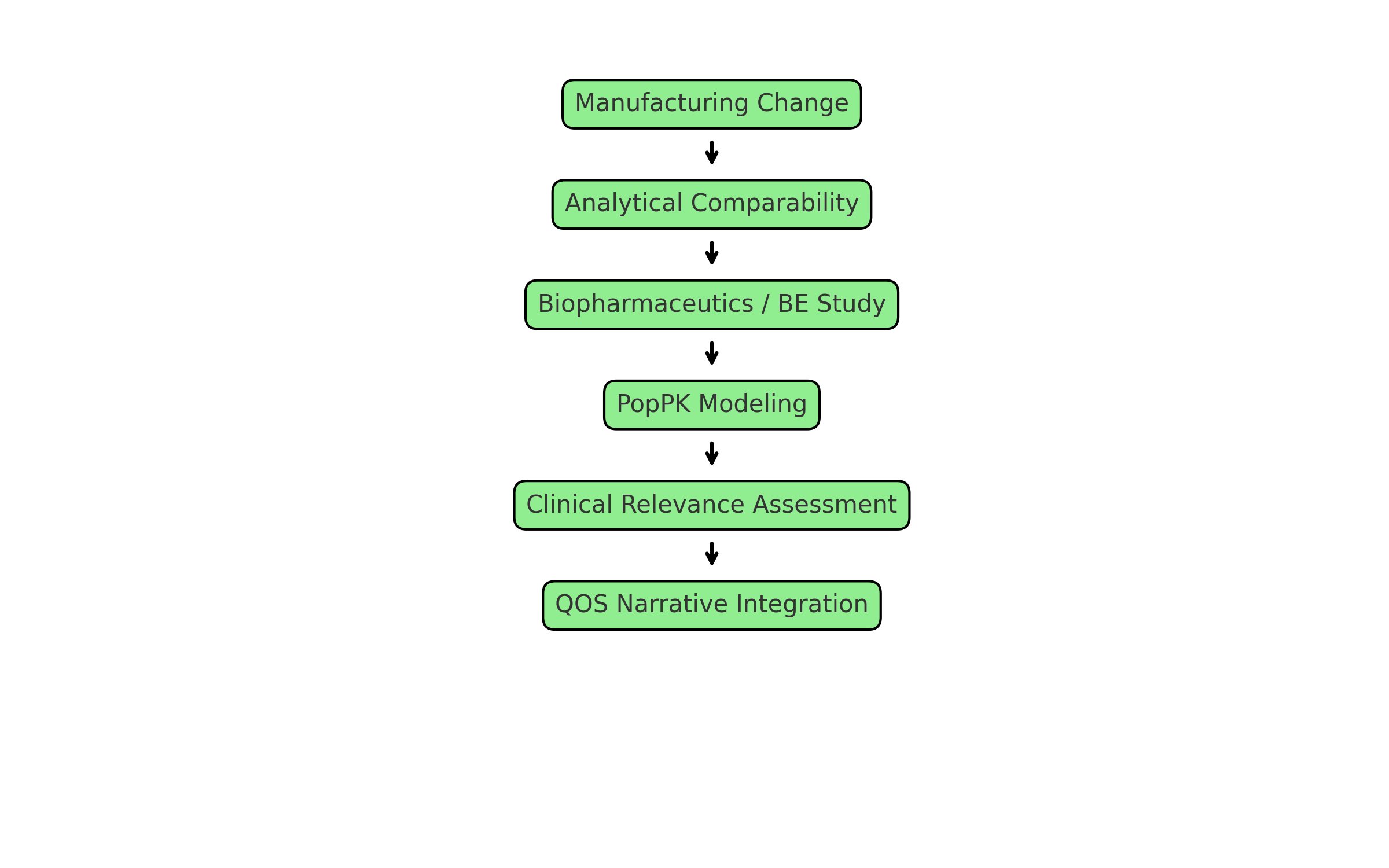
5. Interplay Between CMC and Clinical Pharmacology
- Comparability: Analytical similarity paired with PopPK modeling supports bridging after manufacturing changes.
- Formulation Evolution: Capsule to tablet transitions explained through bioequivalence.
- Specification Setting: Clinical variability drives limits for dissolution, particle size, glycosylation, or DAR.
- Risk-Based Strategy: Quality by design principles are anchored by clinical data.
6. Examples in Practice
To make this concrete, here are mock QOS excerpts (fictitious but modeled on real submissions). These show how CMC, biopharmaceutics, and clinical pharmacology can be integrated.
Example 1: Small Molecule (BCS Class II Oncology Agent)
2.3.P.2 Pharmaceutical Development (Excerpt)
The drug substance is classified as BCS Class II with pH-dependent solubility, leading to dissolution-limited absorption. Early Phase 1 studies used a capsule with micronized API, which showed dose-proportional exposure up to 200 mg. Later studies used a tablet optimized for intestinal dissolution. Comparative bioavailability studies confirmed equivalence, supported by biorelevant dissolution testing.
Population PK across more than 200 subjects showed no clinically relevant differences in exposure between capsule and tablet. This supported a dissolution specification of Q = 80% in 45 minutes at pH 6.8.

Example 2: Monoclonal Antibody
2.3.S.7 Stability (Excerpt)
The antibody shows sensitivity to high-mannose glycoforms, which are associated with faster clearance. Manufacturing specifications for glycosylation were set based on PopPK analyses linking glycan distribution to systemic clearance.
Comparability studies following a cell line change showed analytical similarity. PopPK modeling confirmed that minor shifts in glycosylation profile did not affect clinical exposure, consistent with Phase 1 bridging data.
Example 3: ADC (Antibody-Drug Conjugate)
2.3.P.5 Control of Drug Product (Excerpt)
The ADC has a heterogeneous DAR distribution. Clinical PK analyses showed that high-DAR species contributed disproportionately to exposure and were linked to higher rates of neutropenia.
Manufacturing controls were set to maintain DAR distribution within the clinically acceptable range. PBPK simulations confirmed that the proposed DAR specification (average 3.5-4.0, with ≤15% DAR ≥6) kept exposure and safety consistent with pivotal Phase 2 data.
Example 4: Radiopharmaceutical
2.3.P.8 Stability (Excerpt)
The radiolabeled peptide has a half-life of 6.5 hours, requiring tight control of shelf-life and release testing. Stability studies showed adequate radiochemical purity up to 12 hours post-labeling.
Clinical PK sampling in Phase 1 confirmed clearance consistent with PBPK predictions, and no differences in exposure were observed between fresh and 8-hour-old product. This supports the proposed in-use shelf-life of 8 hours.
7. Regulatory Perspectives
- FDA: Treats QOS as the first-read document, expects alignment with Module 5 (bioequivalence, food-effect, PopPK).
- EMA: Focuses on comparability, especially for biologics and ADCs.
- PMDA: Emphasizes dissolution and integration of bioequivalence.
8. Strategic Takeaways
- Early and ongoing input from clinical pharmacology into the QOS strengthens the narrative and ensures clinical relevance.
- Figures and summary tables are effective for linking product attributes to patient exposure.
- QOS is both a regulatory requirement and a strategic tool.
9. Conclusion
The QOS is not a simple summary of Module 3. It is the CMC story told without rework, concise, clinically anchored, and aligned with the rest of the CTD.
By integrating clinical pharmacology and biopharmaceutics, sponsors can show that specifications are not arbitrary numbers. They are clinically justified guardrails that assure safe, effective, and consistent treatment for patients.