Introduction
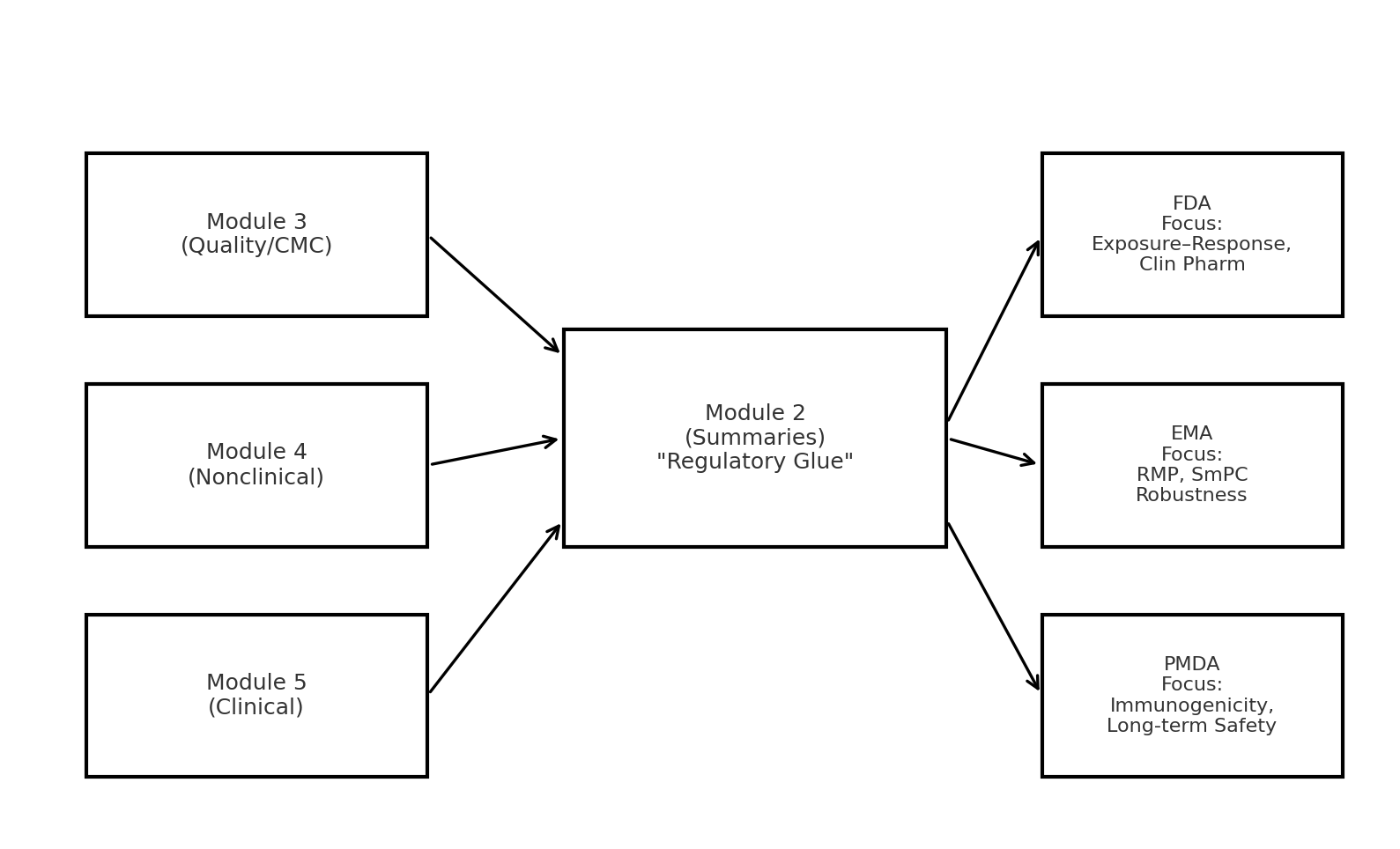
Regulatory submissions succeed or fail not only on the strength of their data but also on how well the story flows across modules. Reviewers are not reading each report in isolation; they start with Module 2 (Summaries) and test whether it coherently reflects the details in Modules 3 (Quality/CMC), 4 (Nonclinical), and 5 (Clinical). Inconsistencies between these modules can undermine trust, while well-aligned narratives can smooth the path to approval.
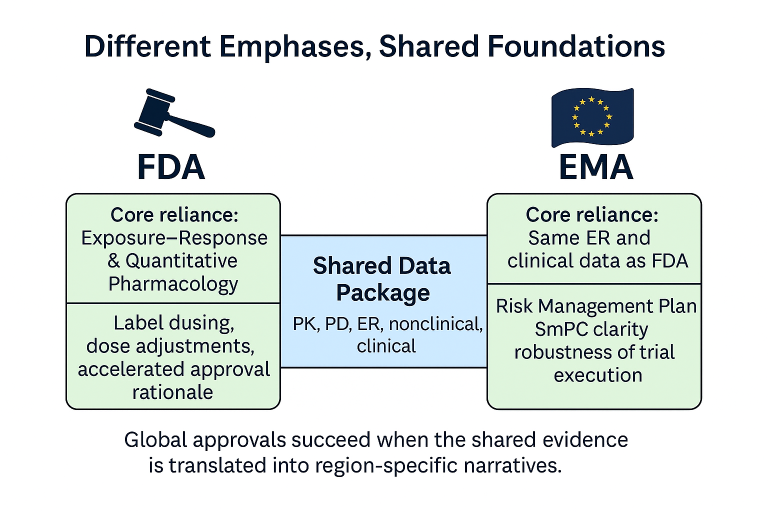
In a globalized industry, submissions are rarely targeted to just one agency [1][2][3]. While the FDA may prioritize exposure–response analyses and quantitative clinical pharmacology [1], the EMA places greater weight on Risk Management Plans (RMPs) [2], clarity in the SmPC [3], and robustness of the evidence. Similarly, the PMDA in Japan often raises specific questions about immunogenicity and long-term safety when reviewing novel therapies. Navigating these differences requires sponsors to treat Module 2 as a true executive summary — a place where the story is tied together for all regulators, not just one.
This article expands on this theme through four detailed case studies — spanning antibody-drug conjugates, RNA therapeutics, small molecules under the Animal Rule, and a classic kinase inhibitor — each illustrating how integration across modules (or lack thereof) shaped regulatory outcomes. We then draw out general recommendations and even sample submission language that can help sponsors avoid pitfalls.
Why Cross-Module Consistency Matters
Module 2 is the “regulatory glue” of the CTD. It is the first stop for regulators and the section they return to when questions arise. Well-crafted Module 2 summaries provide a clear, evidence-based narrative that:
- Weaves together mechanistic rationale, nonclinical findings, and clinical outcomes.
- Signals transparency about limitations while showing how uncertainties are mitigated.
- Provides consistent terminology across the US Prescribing Information (PI) and EU SmPC.
Regional nuances [1][2][3]:
- FDA: Expects detailed exposure–response analyses [1], often asking whether observed safety or efficacy differences are dose- or concentration-driven. ER plots and population PK/PD analyses can tip the balance.
- EMA: Insists that clinical findings be reflected in risk minimisation language and RMP commitments [2]. Even strong efficacy must be contextualized in terms of long-term safety and monitoring.
- PMDA (Japan): Frequently questions immunogenicity in biologics and the long-term translation of nonclinical findings to humans, requiring detailed bridging justifications [13].
“The integrated nonclinical, pharmacology, and clinical data provide a coherent benefit–risk assessment, with uncertainties managed through dose adjustments (US PI), explicit SmPC risk minimisation measures (EU), and ongoing post-marketing surveillance (JP).”
Case 1: Antibody-Drug Conjugate — Gemtuzumab ozogamicin (Mylotarg)
Background: One of the first antibody-drug conjugates, Mylotarg was initially approved in 2000 in the US but later withdrawn due to hepatic veno-occlusive disease (VOD) concerns. It was reintroduced in 2017 with a revised, fractionated dosing regimen.
- FDA: The re-approval was built on ALFA-0701 trial results, supported by integrated nonclinical tox data and exposure–safety analyses [4]. The cross-module integration — showing how dose fractionation aligned with safety improvements — was key to rebuilding trust.
- EMA: Initially refused approval, citing protocol deviations and lack of robust evidence. Later approval (2018) required clear SmPC warnings, a strong RMP, and additional pharmacovigilance commitments [5]. The CHMP also scrutinized the robustness of the trial dataset more heavily than FDA.
Lesson: EMA often goes deeper on trial execution and robustness, whereas FDA may focus on exposure–toxicity tradeoffs. Both demanded clear cross-module consistency.
“Fractionated dosing was prospectively selected to mitigate hepatic toxicity observed in nonclinical and prior clinical studies, while preserving anti-leukemic efficacy. This integrated dataset demonstrates an improved benefit–risk profile.”
Case 2: RNA Therapeutic — Eteplirsen (Exondys 51)
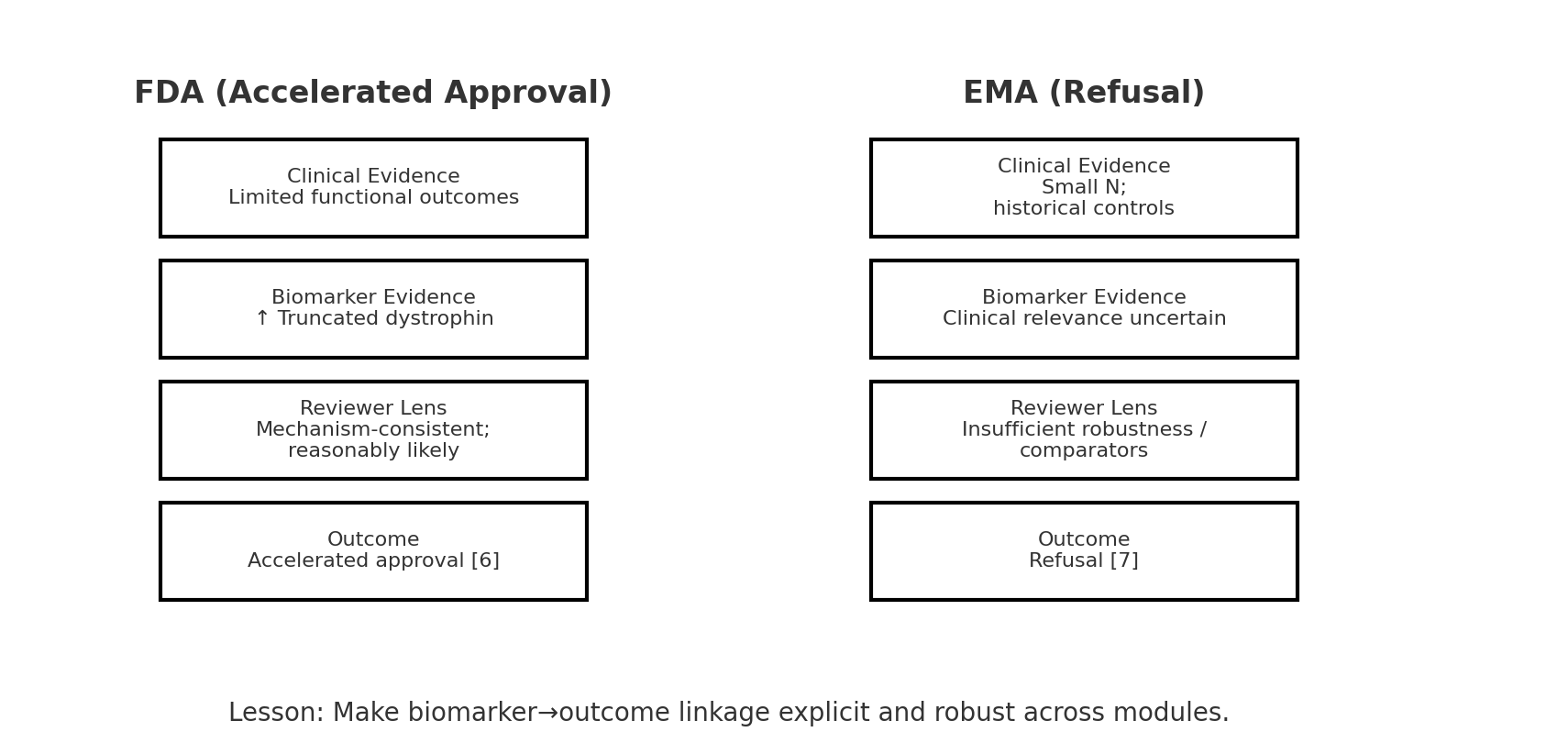
Background: Eteplirsen, an exon-skipping RNA therapeutic for Duchenne muscular dystrophy, illustrates how regional agencies can diverge dramatically.
- FDA: Approved under accelerated approval in 2016, based on truncated dystrophin as a surrogate biomarker [6]. Despite limited functional outcome data, FDA determined the biomarker was “reasonably likely” to predict clinical benefit.
- EMA: CHMP refused marketing authorization in 2018. They cited the very small trial size (~12 patients), reliance on historical controls, and the uncertain clinical relevance of modest dystrophin increases [7]. They explicitly concluded that the cross-module story was not credible.
- PMDA: Japan aligned more closely with EMA, requesting additional confirmatory evidence before considering approval.
Lesson: Biomarker–outcome linkage must be airtight for global acceptance. A submission that relies heavily on surrogates may succeed in the US but fail in the EU.
“While functional benefit has not been conclusively demonstrated, the observed increase in truncated dystrophin is consistent with the drug’s mechanism of action and is reasonably likely to predict clinical benefit.”
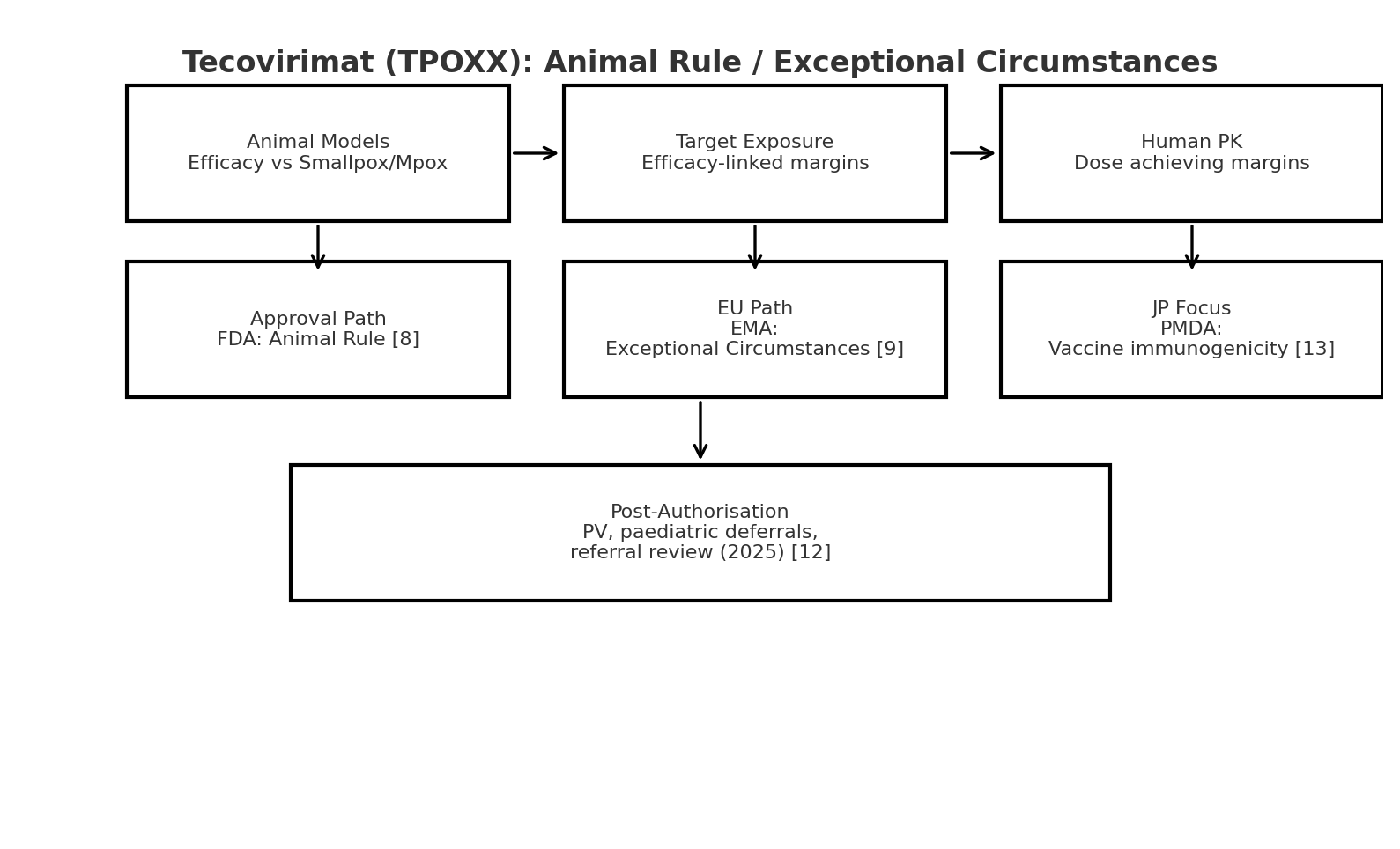
Case 3: Small Molecule under Animal Rule — Tecovirimat (TPOXX)
Background: Developed for smallpox, tecovirimat could not be ethically tested for efficacy in humans. Both FDA and EMA had to rely on animal data bridged to human PK.
- FDA: Approved in 2018 under the Animal Rule [8]. The dose was selected by integrating efficacy data from multiple animal models with human PK exposure margins. Cross-module integration was explicit and central to the submission.
- EMA: Approved in 2022 under “exceptional circumstances.” The EPAR shows how CHMP demanded ongoing post-marketing monitoring, deferred pediatric studies, and extensive risk management commitments [9]. In 2025, EMA initiated a referral review to reassess effectiveness in mpox — showing that the integrative logic must hold up post-approval [12].
- PMDA: Japan also approved tecovirimat but emphasized concerns about vaccine–drug interactions, specifically attenuation of vaccine immunogenicity, and requested ongoing immunogenicity monitoring [13].
Lesson: For novel pathways, agencies will demand that the bridging narrative be crystal clear and resilient to long-term scrutiny.
“Efficacy established in multiple animal models was bridged to humans via pharmacokinetic comparability, with the selected dose providing exposure margins consistent with efficacy in animals.”
Case 4: Classic Exposure–Response Precedent — Sunitinib (Sutent)
Background: A multi-kinase inhibitor widely used in oncology, sunitinib is a positive example of exposure–response (ER) integration done well.
- FDA: FDA reviews emphasized ER analyses that linked dose, exposure, and both efficacy and safety [10]. Label recommendations for interruptions and dose reductions were directly based on ER findings.
- EMA: EPAR and subsequent SmPC updates highlight the importance of clarity in communicating ER-driven dose adjustments [11]. EMA reviewers pushed for precise language so physicians could translate ER science into patient management.
Lesson: Strong ER analyses not only satisfy FDA but also support EMA’s demand for clear, patient-facing SmPC content. This case illustrates how quantitative pharmacology can serve as the ultimate regulatory glue.
Expanded Discussion: Practical Pitfalls and Best Practices
- Data silos kill consistency: Nonclinical and clinical pharmacology teams often work in isolation. Ensure cross-functional review of Module 2 drafts.
- Terminology drift: Inconsistent use of terms like “treatment-emergent adverse event” vs “drug-related AE” across modules confuses reviewers.
- Over-reliance on appendices: Data tables buried in Module 5 with no synthesis in Module 2 weaken the story.
- Ignoring regional expectations: FDA may tolerate exploratory ER analyses, but EMA often demands confirmatory analyses and explicit RMP measures.
- Neglecting post-approval lens: EMA and PMDA frequently revisit approvals (as with tecovirimat). Sponsors should write submissions assuming reviewers will return years later.
General Recommendations for Sponsors
- Write Module 2 as the executive narrative linking all other modules.
- Maintain consistent terminology across PI, SmPC, and clinical summaries.
- Anticipate reviewer cross-checks and explicitly reference supporting modules.
- Always include ER analyses, even exploratory, to strengthen cross-module coherence.
- Address limitations transparently and show how other modules mitigate them.
- Plan globally, not sequentially — align US and EU narratives upfront.
- Use visuals — bridging diagrams and ER plots can convey integration better than text alone.
Conclusion
Cross-module consistency is non-negotiable in regulatory submissions. FDA and EMA may emphasize different aspects—exposure–response vs risk minimisation—but both expect a coherent, aligned story. PMDA and other regulators add further nuances, often focusing on immunogenicity and long-term safety. The case studies of Mylotarg [4][5], Eteplirsen [6][7], Tecovirimat [8][9], and Sunitinib [10][11] show that when the “regulatory glue” holds, challenging programs can succeed globally. When it fails, approvals stall or diverge across regions.
Sponsors that proactively integrate their story, anticipate regional differences, and provide transparent, consistent narratives will improve their chances of smoother global approvals and faster patient access.
References
- U.S. Food and Drug Administration. Guidance for Industry: Exposure–Response Relationships — Study Design, Data Analysis, and Regulatory Applications. 2003. PDF • Web page
- European Medicines Agency. Guideline on Good Pharmacovigilance Practices (GVP) Module V: Risk Management Systems. Rev 2, 2017. PDF • Overview
- European Medicines Agency. Guideline on Summary of Product Characteristics (SmPC). Rev 2, September 2012. Overview • Reference docs
- U.S. Food and Drug Administration. Mylotarg (gemtuzumab ozogamicin) BLA Approval Package, 2017. Approval package PDF • Medical review PDF • Label
- European Medicines Agency. Mylotarg: EPAR – Public assessment report, 2018. EPAR page • Refusal public assessment report (2008)
- U.S. Food and Drug Administration. Exondys 51 (eteplirsen) NDA Approval, 2016. Approval letter PDF • Medical review PDF • Label
- European Medicines Agency. Exondys (eteplirsen): Questions & answers on refusal of the marketing authorisation, 2018. Q&A PDF • EPAR page
- U.S. Food and Drug Administration. TPOXX (tecovirimat) NDA Approval (Animal Rule), 2018. Medical review PDF
- European Medicines Agency. Tecovirimat SIGA: EPAR – Public assessment report, 2022. EPAR PAR PDF
- U.S. Food and Drug Administration. Sutent (sunitinib) NDA Approval Package, 2006. Approval page • Clinical pharmacology/biopharmaceutics review PDF • Medical review PDF
- European Medicines Agency. Sutent: EPAR – Public assessment report. EPAR page
- European Medicines Agency. Tecovirimat SIGA – Referral (Article 20) announcement, July 25, 2025. Referral page
- Pharmaceuticals and Medical Devices Agency (PMDA). Report on the Deliberation Results: Tecovirimat (VP37 inhibitor), December 11, 2024. PDF
Abbreviations
- ADC: Antibody-drug conjugate
- CHMP: Committee for Medicinal Products for Human Use
- CTD: Common Technical Document
- EMA: European Medicines Agency
- EPAR: European Public Assessment Report
- ER: Exposure–response
- FDA: Food and Drug Administration
- MPX: Monkeypox
- PI: Prescribing Information (US)
- PK: Pharmacokinetics
- PMDA: Pharmaceuticals and Medical Devices Agency (Japan)
- RMP: Risk Management Plan
- SmPC: Summary of Product Characteristics
- VOD: Veno-occlusive disease