A joint evolution in FDA and EMA guidance signals a paradigm shift toward efficiency, science, and patient access.
1. The Changing Landscape
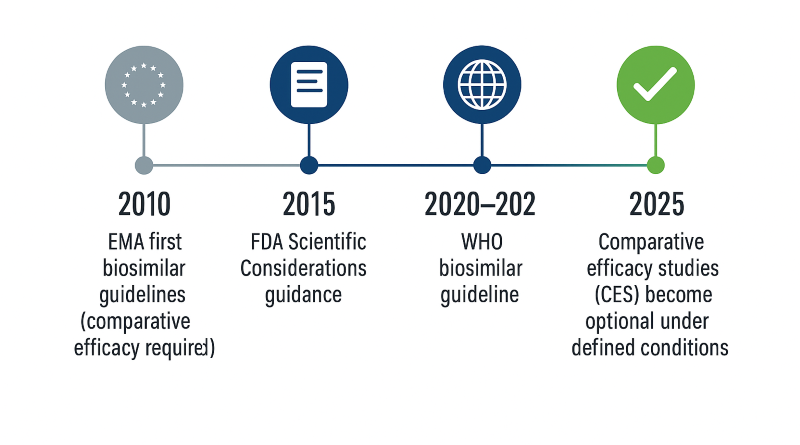
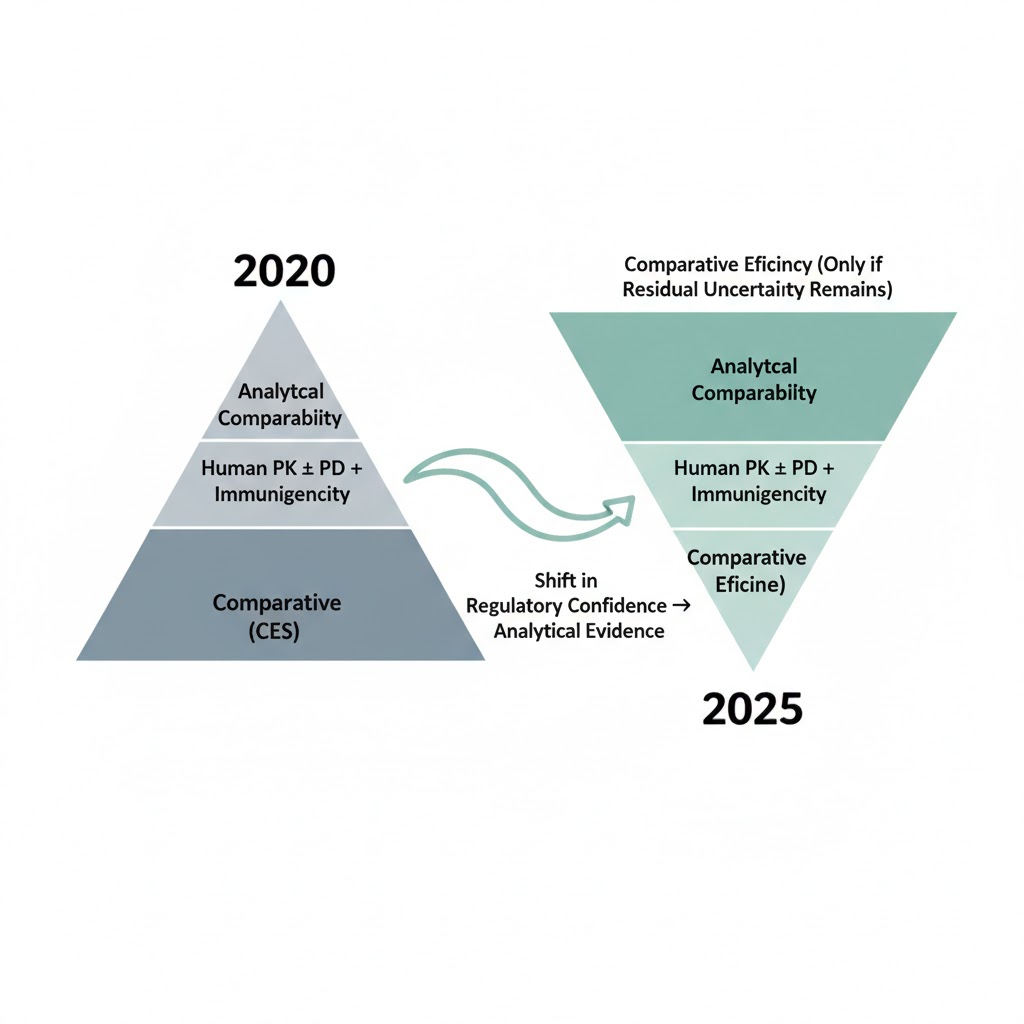
For nearly two decades, biosimilar development followed a familiar path: prove similarity through large, comparative efficacy studies against the reference product. These studies—costly, time-consuming, and often redundant—became the accepted norm. In 2025, both the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) released draft frameworks that fundamentally rethink this model. The new approach shifts emphasis from confirmatory clinical trials to robust analytical and pharmacokinetic (PK) data, acknowledging that modern analytical methods detect differences long before patients ever see them [1,2].
2. The FDA’s Updated Framework
The FDA’s October 2025 Draft Guidance—Scientific Considerations in Demonstrating Biosimilarity to a Reference
Product: Updated Recommendations for Assessing the Need for Comparative Efficacy Studies [1]—formalizes what reviewers have increasingly practiced:
If analytical, PK, and immunogenicity data leave little residual uncertainty, a comparative efficacy study is not scientifically necessary.
- Analytical comparability is the foundation. State-of-the-art biophysical and functional assays (e.g., glycosylation profiles, receptor binding, FcRn binding, potency) are often more sensitive than clinical endpoints [1,3].
- PK and immunogenicity complete the story. Exposure equivalence and comparable immune response are generally sufficient to rule out clinically meaningful differences.
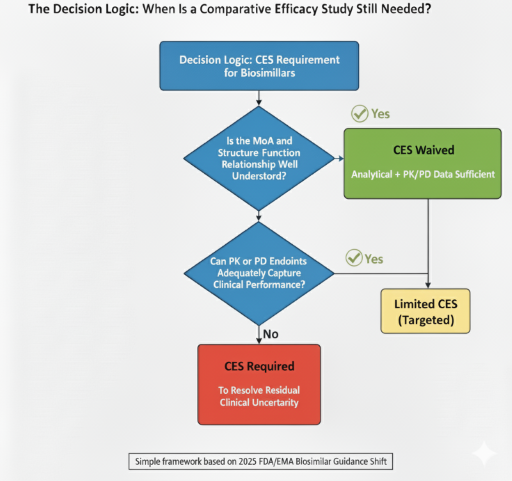
- CES is reserved for special cases. Examples include local-acting products or unclear mechanisms of action.
3. EMA’s Reflection Paper: The Tailored Clinical Approach
The EMA’s March 2025 Reflection Paper on a tailored clinical approach [2] takes a parallel stance, grounded in the principle that structure determines function. If two biologics share indistinguishable structural and functional attributes, comparable clinical efficacy can be inferred [2,4].
Prerequisites to waive CES:
- Mechanism of Action (MoA) is well understood.
- Extensive analytical comparability using orthogonal assays confirms functional equivalence.
- A human PK study demonstrates comparable exposure and immunogenicity.
- Manufacturing consistency is assured through validated controls.
4. The New Hierarchy of Evidence and Clinical Requirements
4.1 Pharmacokinetic (PK) Requirements
Although designs resemble those used for generic drugs, the intent is broader: confirm no clinically meaningful difference in exposure and variability, not just mathematical equivalence.
FDA (2025 Draft) [1]:
- Typically one appropriately designed PK study; parallel or crossover based on half-life and immunogenicity risk.
- Primary metrics: AUC0–t, AUC0–∞, Cmax.
- Acceptance: 90% CI for the GMR within 80–125%; interpreted within the totality-of-evidence.
- Conduct in the most sensitive population (often HVs if ethical/feasible).
EMA (Reflection Paper) [2]:
- Comparative PK is the pivotal clinical study; single-dose design usually sufficient in a sensitive population.
- PD endpoints can supplement PK when mechanistically relevant (e.g., G-CSF, insulin).
- 80–125% CI remains conventional; scientifically justified alternatives allowed for highly variable biologics.
Contrast with generics: Small-molecule BE focuses strictly on absorption with the 80–125% rule applied as a hard criterion; biosimilar regulators judge whether any deviation is clinically irrelevant in the context of analytical and mechanistic data [5].
4.2 Immunogenicity Requirements
Immunogenicity assessment distinguishes biosimilars from generics.
FDA [1]:
- Comparative immunogenicity is expected unless a science-based waiver is justified.
- Randomized, parallel-group design comparing biosimilar vs reference; duration covers antibody formation and plateau.
- Endpoints: ADA incidence/titers and NAb; PK analyzed overall and stratified by ADA status.
EMA [2]:
- Comparative immunogenicity required—often integrated with PK; waiver only with strong analytical fingerprint and extensive class experience.
- Interpretation is comparative rather than based on absolute thresholds.
Generics: Small-molecule generics are exempt because they lack protein epitopes [5].
4.3 Summary of Comparative Requirements
| Parameter | Biosimilars (2025) | Generics |
|---|---|---|
| Analytical similarity | Mandatory and decisive | Not required |
| PK design | Comparative; single-dose; parallel or crossover | Two-way crossover |
| PK acceptance range | 80–125% (contextual, totality-of-evidence) | 80–125% (strict) |
| PD | Optional, if relevant | Rare |
| Immunogenicity | Required unless waived | Not applicable |
| Comparator | Licensed reference biologic | Reference drug |
| Interpretation | “No clinically meaningful difference” | “Identical exposure” |
5. Key Differences Between FDA and EMA Approaches
| Aspect | FDA (2025 Draft Guidance) | EMA (2025 Reflection Paper) |
|---|---|---|
| Regulatory form | Guidance for industry | Reflection paper (pre-guideline) |
| Core principle | CES may not be necessary | Analytical + PK may be sufficient |
| Tone | Flexible, case-specific | Structured, science-based |
| Scope | Therapeutic proteins under 351(k) | All biotech-derived proteins |
| Residual uncertainty | Discussed early with FDA | Quantified via risk-based matrix |
| Terminology | “Streamlined approach” | “Tailored clinical approach” |
6. Development Impact: Faster, Leaner, More Predictable

7. When Is a Comparative Efficacy Study Still Needed?
A CES is now reserved for situations where uncertainty remains—typically local-acting products (e.g., ophthalmic or inhaled) or molecules with unclear structure–function relationships [1,2]. Developers should present a transparent decision logic to justify any clinical endpoint study.
8. What This Means for Developers
- Earlier clarity on program scope driven by analytics and PK.
- Fewer redundant trials and lower capital risk.
- More predictable interactions with FDA/EMA based on quantitative criteria.
9. Conclusion – Analytical Confidence Defines the Future
FDA and EMA now recognize that analytical and PK data, interpreted under the totality-of-evidence paradigm, are the most sensitive indicators of biosimilarity [1–4]. Regulatory confidence stems from molecular precision—not trial size.
10. Partnering for a Smarter Biosimilar Strategy
At ClinPharm Dev Solutions, we design biosimilar programs built on analytical excellence, optimized PK/PD design, and data-driven regulatory strategy. Whether you need a CES waiver justification, a similarity risk assessment, or FDA/EMA scientific advice packages, we can accelerate your path to approval. Contact us to discuss your program.
References
- FDA. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product: Updated Recommendations for Assessing the Need for Comparative Efficacy Studies. Draft Guidance for Industry. October 2025.
- EMA. Reflection Paper on a Tailored Clinical Approach in Biosimilar Development (EMA/CHMP/BMWP/60916/2025). March 2025.
- Cavazzoni P., Yim S. The Science of Biosimilars – Updating Interchangeability. JAMA. 2024;332(15):1235–1236.
- Kirsch-Stefan N., Guillen E., Ekman N., et al. Do the Outcomes of Clinical Efficacy Trials Matter in Regulatory Decision-Making for Biosimilars? BioDrugs. 2023;37(6):855–871.
- FDA. Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted Under an ANDA. Guidance for Industry. December 2022.
- Bielsky M.C., et al. Streamlined Approval of Biosimilars: Moving on from the Confirmatory Efficacy Trial. Drug Discovery Today. 2020;25(11):1910–1918.
Abbreviations
ADA – Anti-Drug Antibody
AUC – Area Under the Concentration–Time Curve
BLA – Biologics License Application
CES – Comparative Efficacy Study
CI – Confidence Interval
CQAs – Critical Quality Attributes
EMA – European Medicines Agency
FDA – Food and Drug Administration
GMR – Geometric Mean Ratio
MoA – Mechanism of Action
NAb – Neutralizing Antibody
PD – Pharmacodynamics
PK – Pharmacokinetics
PHS – Public Health Service
WHO – World Health Organization