Executive Outline: Series on Modality-Specific Clinical Pharmacology
To address the rapidly evolving landscape of therapeutic modalities and the increasing complexity of regulatory requirements, this comprehensive report serves as the foundational document in a specialized series detailing modality-specific requirements for clinical pharmacology, drug metabolism and pharmacokinetics (DMPK), and pharmacometrics. The series is structured to provide an exhaustive analysis of how varying therapeutic architectures dictate specialized developmental strategies.
| Article Sequence | Thematic Focus | Core Topics Addressed |
|---|---|---|
| Article 1 (Current Report) | Overview of Modalities and the Clinical Pharmacology Plan | Integration of DMPK and pharmacometrics; regulatory paradigms; foundational principles for small molecules, monoclonal antibodies, bispecifics, antibody-drug conjugates, and radiopharmaceuticals. |
| Article 2 | Small Molecule Pharmacokinetics and Bioavailability | Biopharmaceutics Classification System (BCS); formulation strategies; mechanistic food effect modeling; intricate hepatic-renal interplay and uremic toxin modulation. |
| Article 3 | Large Molecule Biologics and Subcutaneous Delivery | Monoclonal antibody disposition; lymphatic absorption barriers; target-mediated drug disposition (TMDD); time-varying clearance mechanisms; immunogenicity profiling. |
| Article 4 | Complex Biologicals: Bispecifics and Conjugates | Dual-target engagement pharmacometrics; trimeric complex modeling; multi-analyte bioanalytical strategies; payload-driven drug-drug interactions (DDIs); QTc liabilities. |
| Article 5 | Radiopharmaceuticals and Theranostics | Medical Internal Radiation Dose (MIRD) formalism; biological versus physical half-life; dosimetry; radiation safety operations; regulatory frameworks (FDA/Euratom). |
Introduction: The Evolution and Mandate of the Clinical Pharmacology Plan
The paradigm of pharmaceutical drug development has undergone a profound transformation, moving away from empirical, trial-and-error dose-finding toward a highly mechanistic, model-informed discipline. At the absolute core of this scientific evolution is the Clinical Pharmacology Plan (CPP), an exhaustive, multidisciplinary framework designed to delineate the precise dose-concentration-effect relationship of an investigational therapeutic agent across diverse human populations.
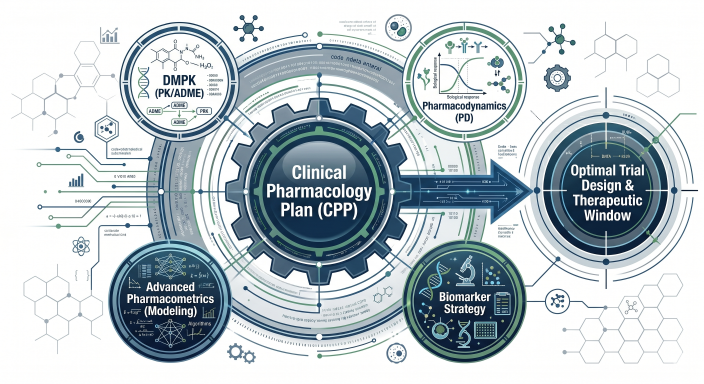
Figure 1: The Anatomy of a Modern Clinical Pharmacology Plan (CPP)
The CPP serves as the strategic operational blueprint for the entire clinical development lifecycle, encompassing the interdependent disciplines of drug metabolism and pharmacokinetics (DMPK), pharmacodynamics (PD), and advanced pharmacometrics. By continuously integrating physiological, biological, and mathematical modeling, the CPP ensures the optimal design of clinical trial protocols, establishes robust therapeutic windows, and proactively addresses the unique pharmacokinetic and pharmacodynamic alterations present in specialized populations, such as pediatric, geriatric, and organ-impaired cohorts.
Modern therapeutic development relies heavily on the rigorous application of fundamental pharmacokinetic and pharmacodynamic principles to successfully navigate the high-risk transition from first-in-human (FIH) studies to pivotal phase III trials. The foundational principles of clinical pharmacology dictate that an investigational candidate’s survival in advanced clinical trials is completely contingent upon the validation of three critical pillars: verifiable drug exposure at the intended site of action, demonstrable target binding within the relevant biological matrix, and the explicit expression of functional pharmacological activity. These criteria are collectively termed the “three pillars of survival.”
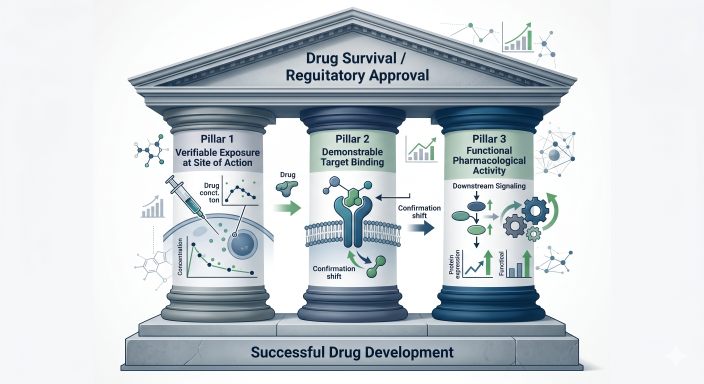
Figure 2: The Three Pillars of Survival in Drug Development
A failure to accurately characterize and validate any of these pillars significantly elevates the risk of late-stage attrition, resulting in catastrophic losses of resources and delayed patient access to critical therapies. Consequently, global regulatory authorities, including the United States Food and Drug Administration (FDA) Office of Clinical Pharmacology and the European Medicines Agency (EMA), stringently mandate the early, continuous, and exhaustive use of quantitative clinical pharmacology to justify development decisions.
The Role of Pharmacometrics and Model-Informed Drug Development
Pharmacometrics—the quantitative science of utilizing mathematical and statistical models to characterize, understand, and predict a drug’s pharmacokinetic and pharmacodynamic behavior—serves as the analytical engine of the modern CPP. Utilizing sophisticated computational techniques such as population pharmacokinetics (PopPK), physiologically based pharmacokinetic (PBPK) modeling, and complex exposure-response analysis, pharmacometrics enables developers to simulate hypothetical clinical scenarios, predict unstudied dose levels, and derive rational starting doses for FIH trials.
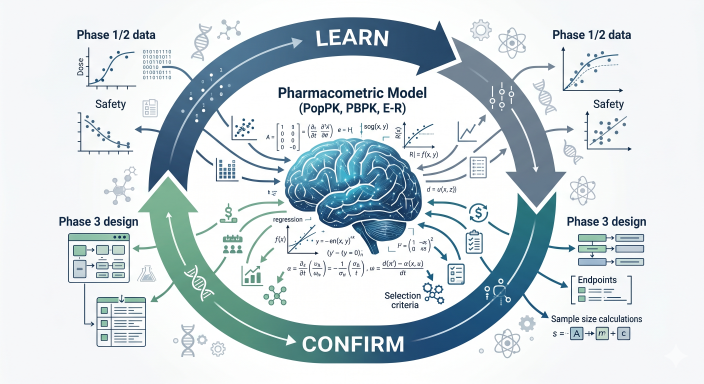
Figure 3: The Model-Informed Drug Development (MIDD) Iterative Workflow
The FDA’s Division of Pharmacometrics heavily emphasizes that regulatory decisions concerning the efficiency of drug development, ultimate approval, and product labeling are driven by the wisdom derived from these quantitative models. Drug development requires several iterations of pharmacometric model-informed learning and confirming. This iterative process includes modeling to deeply understand the dose-response relationship in preclinical animal models, deriving a safe maximum starting dose for human administration, and conducting the overall analysis of Phase I and Phase II data to mathematically optimize the dose for safety and efficacy in Phase III pivotal trials. Furthermore, pharmacometrics extends beyond regulatory approval, assisting clinical pharmacists and physicians in clinical practice by determining off-label dosing in special populations and individualizing dosing based on measured biomarkers for personalized medicine.
Small Molecules: Classic Pharmacokinetics and Bioavailability Optimization
Small molecule therapeutics remain the foundational pillar of systemic pharmacotherapy. Constructing a comprehensive CPP for a small molecule requires the meticulous evaluation of its absorption, distribution, metabolism, and excretion (ADME), with a particular emphasis on oral bioavailability, intrinsic physiological factors, and high susceptibility to food effects.
Bioavailability and the Biopharmaceutics Classification System
The oral bioavailability of a small molecule is dictated by a complex, dynamic interplay of its inherent physicochemical properties, human gastrointestinal physiology, and pre-systemic hepatic metabolism. The Biopharmaceutics Classification System (BCS) provides a vital framework for guiding formulation strategies. Highly soluble and highly permeable compounds (Class I) generally exhibit excellent bioavailability. Conversely, compounds suffering from poor aqueous solubility (Class II) or poor intestinal permeability (Class III) present substantial challenges.
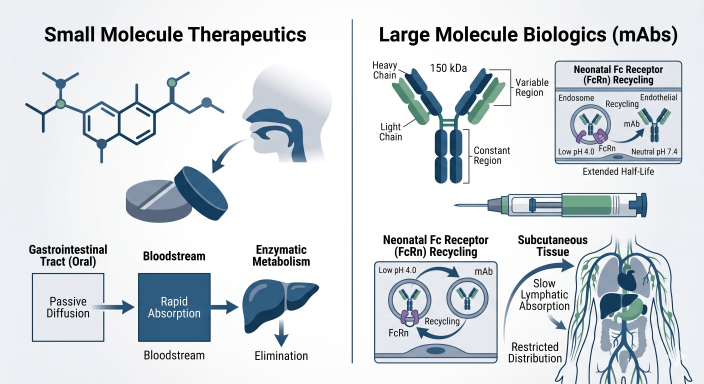
Figure 4: The BCS Matrix and Strategic Formulation Pathways
| BCS Class | Pharmacokinetic Characteristics | Typical Formulation Strategy |
|---|---|---|
| Class I | High Solubility, High Permeability | Standard IR tablets/capsules; biowaivers. |
| Class IIa | Low Solubility (Dissolution limited), High Permeability | Particle size reduction (micronization/nanomilling). |
| Class IIb | Low Solubility (Absolute limited), High Permeability | Amorphous solid dispersions, hot-melt extrusion. |
| Class III | High Solubility, Low Permeability | Permeation enhancers, lipid-based formulations. |
| Class IV | Low Solubility, Low Permeability | Combination of solubility and permeability enhancement. |
Food Effect and Gastrointestinal Physiology Modeling
The administration of a drug product concurrent with food ingestion can significantly alter its systemic availability. Molecules characterized by short elimination half-lives and extensive first-pass hepatic metabolism are particularly susceptible to clinically significant food effects. Advanced PBPK modeling is frequently deployed during early clinical development to simulate and predict food effects before conducting expensive clinical trials.
Hepatic and Renal Impairment Dynamics
The assessment of intrinsic factors is a critical regulatory requirement. Cross-organ communication networks, such as the accumulation of uremic toxins secondary to renal failure modifying the activity of hepatic transporters, must be accounted for. The clinical development strategy must assume that severe renal impairment may compromise both renal clearance and hepatic metabolic capacity.
Monoclonal Antibodies: The Fundamentals of Biologic Disposition
Monoclonal antibodies (mAbs) represent a fundamental paradigm shift. Their clinical pharmacology is defined by restricted biodistribution, protection from catabolism via neonatal Fc receptor (FcRn) recycling, and complex non-linear clearance mechanisms.
Absorption Barriers and Subcutaneous Bioavailability
While intravenous (IV) infusion guarantees systemic bioavailability, the landscape is shifting toward subcutaneous (SC) administration. SC delivery introduces pharmacokinetic complexities, including incomplete bioavailability and delayed time to peak concentration ($T_{max}$) due to lymphatic drainage reliance.
Target-Mediated Drug Disposition and Time-Varying Clearance
The elimination of mAbs is often overshadowed by Target-Mediated Drug Disposition (TMDD), introducing non-linearity into the pharmacokinetic profile. In oncology mAbs, time-varying clearance is an additional challenge; as tumor burden decreases, the systemic hyper-catabolic state normalizes, reducing the antibody’s non-specific degradation rate.
Bispecific Antibodies: Dual-Target Complexity and Spatial Pharmacometrics
Bispecific antibodies (bsAbs) engage two antigens, facilitating novel mechanisms like physically linking T-cells to tumor cells. Pharmacokinetic behavior is exceptionally complex due to simultaneous presence of multiple targets with differing biological properties.
Clinical pharmacologists employ advanced mPBPK models to simulate binding kinetics to both targets. Models help clinical teams avoid the “bell-shaped” dose-response curve where overly high concentrations paradoxically abolish cellular cross-linking efficacy.
Regulatory Considerations and the MABEL Approach
Initial FIH dose selection for bsAbs must utilize a Minimum Anticipated Biological Effect Level (MABEL) approach, setting the dose based on the lowest concentration required to trigger target engagement in vitro, mitigating the risk of cytokine release syndrome.
Antibody-Drug Conjugates: The Trimodal Analytical Challenge
An ADC is a tripartite molecule comprising a monoclonal antibody, a cytotoxic payload, and a chemical linker. This sophisticated design maximizes target cell death while minimizing systemic exposure to healthy tissues.
Multi-Analyte Bioanalytical Strategy and Drug-Drug Interactions
Pharmacokinetic assays must independently quantify three circulating entities: the intact ADC, the total antibody, and the unconjugated payload. Furthermore, because the unconjugated payload is a highly potent small molecule, ADCs inherit DDI liabilities and require rigorous QTc prolongation assessments.
Radiopharmaceuticals: Dosimetry, MIRD, and Radiation Pharmacokinetics
Radiopharmaceuticals consist of a targeting vector bound to a radioactive isotope. The intersection of pharmacological disposition and physical radioactive decay necessitates a CPP built entirely around advanced radiation dosimetry and the Medical Internal Radiation Dose (MIRD) schema.
Dosage Optimization and Theranostics
“Theranostics” utilizes matched pairs of radioisotopes (e.g., Ga-68 for mapping and Lu-177 for treatment). The diagnostic agent acts as an individualized pharmacokinetic probe, allowing clinicians to calculate the maximum therapeutic radiation dose a patient’s organs can tolerate.
Comparative Modality Requirements in Clinical Pharmacology
| Parameter | Small Molecules | mAbs | Bispecifics & ADCs | Radiopharmaceuticals |
|---|---|---|---|---|
| Clearance | Hepatic metabolism; Renal transporters. | TMDD; Lysosomal catabolism; FcRn. | Complex: TMDD (Intact); Hepatic (Payload). | Vector clearance + physical decay. |
| Immunogenicity | Very Low / Rare. | Moderate to High. | Very High. Multi-tiered assays. | Dependent on vector. |
| DDI Liability | High (Direct substrates). | Low (Cytokine mediated). | High (Payload/Cytokine). | Minimal pharmacologic DDI. |
| Organ Strategy | Early renal/hepatic studies. | Generally negligible impact. | Extremely High scrutiny. | High (Excretion pathways). |
Conclusion: The Convergence of Modalities and Pharmacometrics
As pharmaceutical drug development progresses into the future of precision medicine, the rigid boundaries between these modalities are increasingly overlapping. Model-Informed Drug Development has become the universal thread connecting these technologies. By proactively embedding mechanistic physiological considerations into the CPP foundation, developers can mitigate the risk of late-stage failure and accelerate the delivery of life-saving therapeutics.
Abbreviations
ACAT: Advanced Compartmental Absorption and Transit
ADA: Anti-Drug Antibody
ADC: Antibody-Drug Conjugate
ADME: Absorption, Distribution, Metabolism, Excretion
BCS: Biopharmaceutics Classification System
BDDCS: Biopharmaceutics Drug Disposition Classification System
bsAb: Bispecific Antibody
CPP: Clinical Pharmacology Plan
CYP: Cytochrome P450
DDI: Drug-Drug Interaction
DMPK: Drug Metabolism and Pharmacokinetics
FcRn: Neonatal Fc Receptor
FIH: First-in-Human
MABEL: Minimum Anticipated Biological Effect Level
mPBPK: minimal Physiologically Based Pharmacokinetic
MIRD: Medical Internal Radiation Dose
nAb: Neutralizing Antibody
PBPK: Physiologically Based Pharmacokinetic
PopPK: Population Pharmacokinetics
QSP: Quantitative Systems Pharmacology
TMDD: Target-Mediated Drug Disposition
Technical References
- FDA Guidance for Industry (2024). Clinical Pharmacology Considerations for Antibody-Drug Conjugates.
- FDA Guidance for Industry (2026). Pharmacometrics in Regulatory Submissions: Model-Informed Drug Development.
- EMA Guideline (2025). Clinical Pharmacology and Pharmacokinetics of Monoclonal Antibodies.
- Sheiner, L. B., & Beal, S. L. (1980). Evaluation of methods for estimating population pharmacokinetic parameters. Journal of Pharmacokinetics and Biopharmaceutics.
- Amidon, G. L., et al. (1995). A theoretical basis for a biopharmaceutic drug classification. Pharmaceutical Research.
- Sadekar, S., et al. (2022). Clinical Pharmacology Considerations for Bispecific Antibodies. Clinical Pharmacology & Therapeutics.
- Zuckier, L. S., et al. (2020). Principles of Radiopharmaceutical Dosimetry and the MIRD Formalism. Journal of Nuclear Medicine.
- Project Optimus (FDA Oncology Center of Excellence, 2026). Dose Optimization for Oncology Drug Development.