Beyond Biosimilarity: The 2026 Strategic Blueprint for Day 1 Interchangeability Executive Summary For over a decade, the Interchangeable designation was the most expensive badge of honor in the biosimilar industry, typically requiring a multi-million dollar clinical switching trial. As of early 2026, the regulatory wall has collapsed. Following the FDA’s pivotal June 2024 guidance and […]
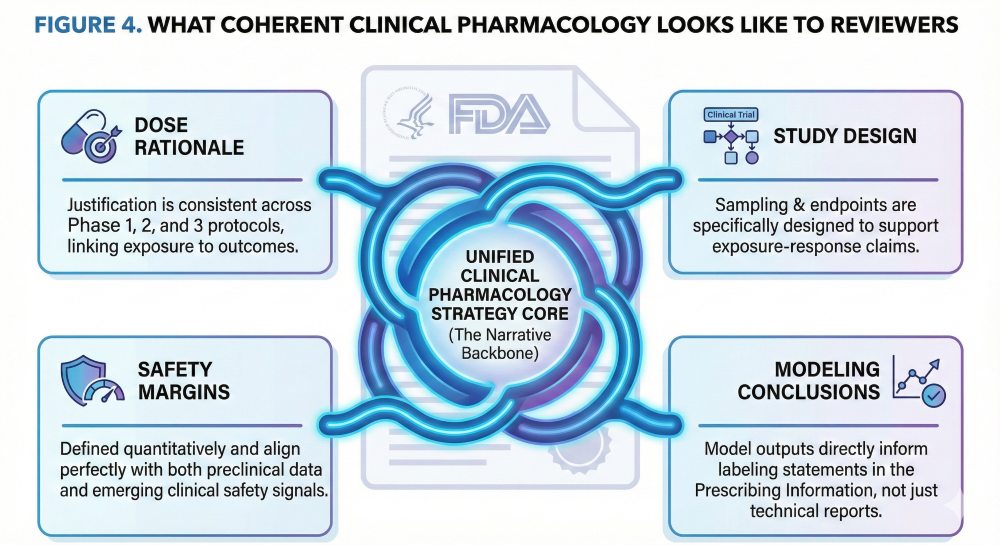
The Quiet Risk in 2026 INDs: When Clinical Pharmacology Is Treated as a Deliverable Instead of a Strategy
Many 2026 INDs are already being scoped. Budgets are being finalized, CROs are being shortlisted, and timelines are being compressed. In that environment, clinical pharmacology often becomes something to be delivered rather than something to be designed. Analyses are commissioned because the IND needs them, not because a decision requires them. The risk here is
Strategic Efficiency: How Model-Informed Drug Development (MIDD) Can Waive Clinical Studies and Accelerate Timelines
Introduction: The High Cost of “Checking the Box” For emerging biotech companies, cash runway is oxygen. The cost of developing a new drug has skyrocketed to over $2 billion, with the clinical phase consuming the vast majority of that budget. Every month spent in clinical development burns capital, shortens patent life, and delays the path
A Practical Guide to Quantitative Pharmacology in Drug Development
When to Use PopPK/PD, Exposure Response, PBPK, and QSP, and When Not To 1. Introduction: Why Quantitative Pharmacology Matters Model informed development has shifted from optional exploratory analysis to a central part of decision making. Most high value decisions such as target viability, mechanism plausibility, first in human (FIH) dose selection, Phase 2 dose justification,
A Practical Guide to Quantitative Pharmacology in Drug Development Read More »
Modeling & Simulation as Regulatory Evidence in the CTD: Practical Case Studies Across PopPK/PD, PBPK, E–R, and QSP
Model-informed analyses are increasingly recognized by regulators and sponsors as credible evidence—especially when the question is clear, the model is verified for purpose, and uncertainties are transparent. This article reviews real submissions where M&S supported dose selection, regimen bridging, DDI labeling, pediatric dosing, and systems-level benefit–risk insight. Contents Introduction Case 1 — Exposure Bridging (Pembrolizumab
Biosimilar Development in 2025: From Comparative Trials to Analytical Confidence
A joint evolution in FDA and EMA guidance signals a paradigm shift toward efficiency, science, and patient access. 1. The Changing Landscape For nearly two decades, biosimilar development followed a familiar path: prove similarity through large, comparative efficacy studies against the reference product. These studies—costly, time-consuming, and often redundant—became the accepted norm. In 2025, both
Biosimilar Development in 2025: From Comparative Trials to Analytical Confidence Read More »
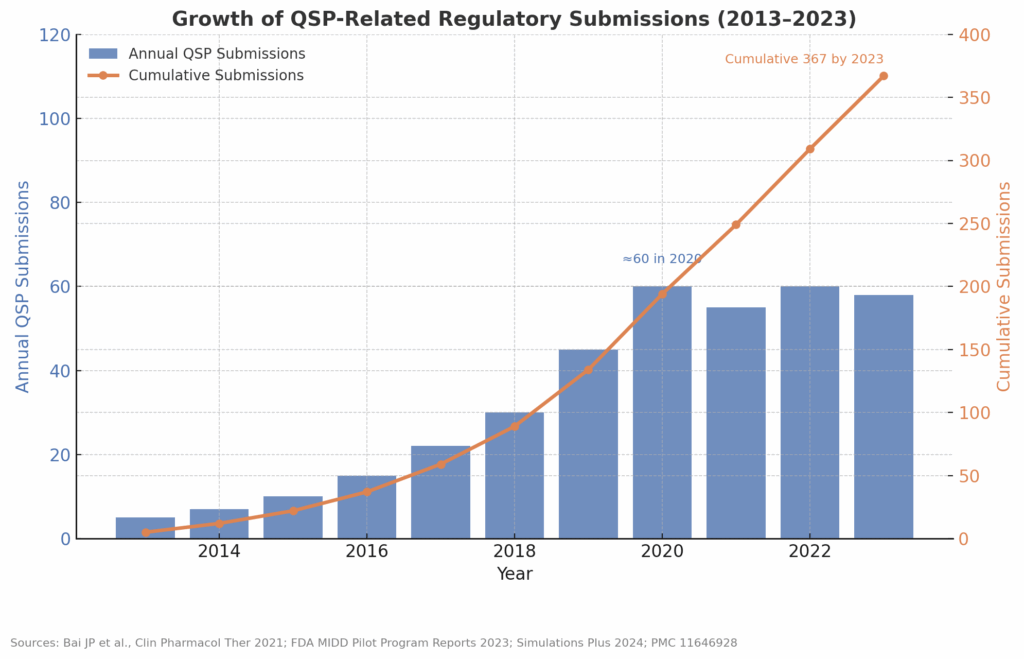
Quantitative Systems Pharmacology (QSP) in Clinical Development: When to Use It, What It Delivers, and How to Make It Regulatory-Ready
Audience: biotech C-suites, program leaders, clinical pharmacology & modeling teams. Why QSP matters now Sponsors increasingly face decisions where empirical models alone (PopPK, exposure–response) are not enough—novel mechanisms, combinations, complex biology, and heterogeneous populations. Quantitative Systems Pharmacology (QSP) integrates disease/biology knowledge with pharmacology to generate testable, decision-grade predictions across the translational and clinical continuum (e.g.,
AI as a Scientific Partner: Where Large Language Models Really Fit in Drug Development
By Aniruddha Amrite, ClinPharm Dev Solutions LLC Executive summary (for busy leaders) Large language models (LLMs) are already useful across the drug development continuum—if we anchor them to trusted sources and keep humans in the loop. Practical wins today include faster literature synthesis and hypothesis triage, eligibility criteria parsing and patient–trial matching at scale, drafting
AI as a Scientific Partner: Where Large Language Models Really Fit in Drug Development Read More »
The Regulatory Glue: Integrating Modules 2–5 for FDA, EMA, and Beyond
Figure 1. Global “regulatory glue” overview: a shared evidence package (PK/PD, ER, nonclinical, clinical) flows into Module 2 and is translated for multiple agencies. Introduction Regulatory submissions succeed or fail not only on the strength of their data but also on how well the story flows across modules. Reviewers are not reading each report in
The Regulatory Glue: Integrating Modules 2–5 for FDA, EMA, and Beyond Read More »
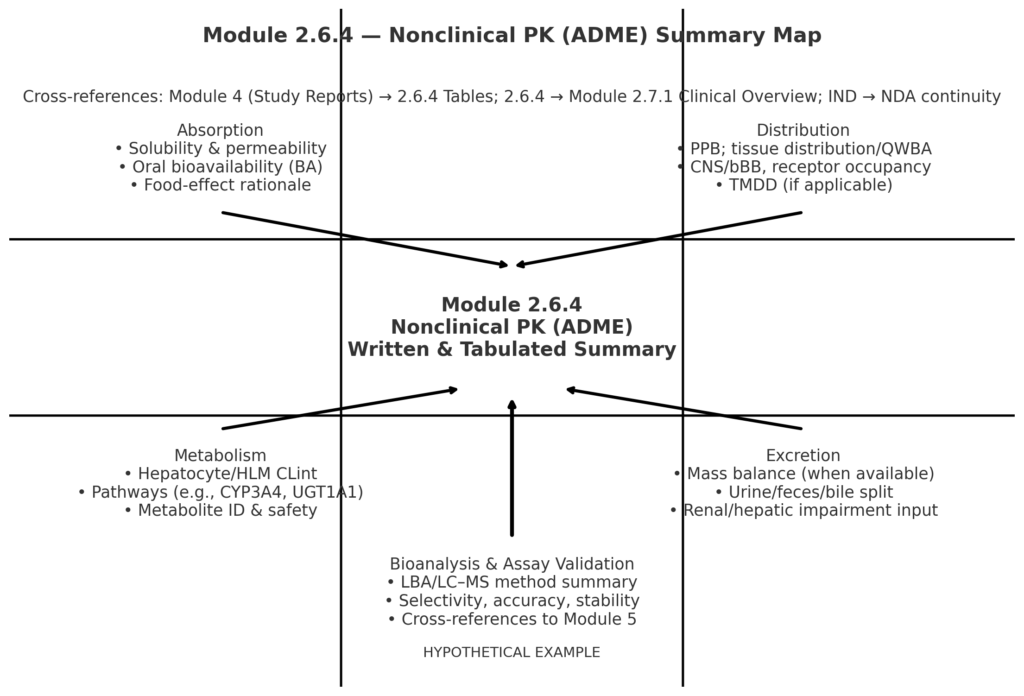
CTD 2.6.4 Playbook: Writing a Reviewer-Ready Nonclinical ADME That Bridges Module 4 to 2.7.1 (with Modality-Specific Examples)
Module 2.6.4 is the reviewer’s fast path to your nonclinical ADME story. Done well, it shows what was studied, what was learned, and why those data are sufficient for the current stage of development and the proposed clinical plan, with traceability back to Module 4 and forward to the clinical overview (2.7.1) [1]. What belongs