Executive Summary
For a long time, the pharmaceutical industry viewed Clinical Pharmacology models as helpful supporting characters. They were tools used to guide a trial design or help justify a specific dose. As we move through 2026, a fundamental shift has taken place. The FDA and EMA have moved from Model Informed to Model Integrated Evidence (MIE). In this new reality, the model is no longer just a consultant: it is the primary evidence [1, 2]. For sponsors working in rare diseases, pediatric populations, or complex drug interactions, MIE is the ultimate regulatory biowaiver.
1. Moving Beyond Informed to Integrated
Distinguishing between MIDD and MIE might feel like splitting hairs, but it is actually a major shift in how the FDA views your data. While MIDD uses models to guide clinical decisions, MIE uses models to generate the actual data that appears on the drug label [2]. This transition has been picking up speed thanks to the success of the PDUFA VII commitments and the finalized 2025 frameworks on model integration [1].
The regulatory question in 2026 has evolved. It is no longer about whether the model supports your clinical findings. It is now about whether the model is robust enough to stand in place of a clinical finding. This allows you to bypass the traditional trial and error approach, especially when recruitment is a bottleneck or ethical constraints make large scale testing impossible.
 Figure 1: The Evolution of Evidence. Moving from traditional clinical observations to Model-Integrated Evidence (MIE).
Figure 1: The Evolution of Evidence. Moving from traditional clinical observations to Model-Integrated Evidence (MIE).
2. The Regulatory Turning Point
The pivot toward MIE was cemented by a series of landmark approvals where clinical trials for secondary indications were waived entirely. The 2024 draft guidance on the use of PBPK modeling for pediatric extrapolation was the first real catalyst [5]. By 2025, the FDA demonstrated that for certain low uncertainty questions, like drug interactions and organ impairment, simulation was statistically more reliable than small, high variability clinical groups [1, 2].
3. Where MIE is Winning Right Now
Pediatric and Rare Disease Extrapolation
In populations where the total number of patients is limited, every person is a precious resource. MIE allows you to leverage adult data through PBPK and Population PK models to define pediatric dosing with high confidence [5]. This often eliminates the need for Phase 3 efficacy studies in children, letting you move straight from adult approval to a pediatric label extension [4].
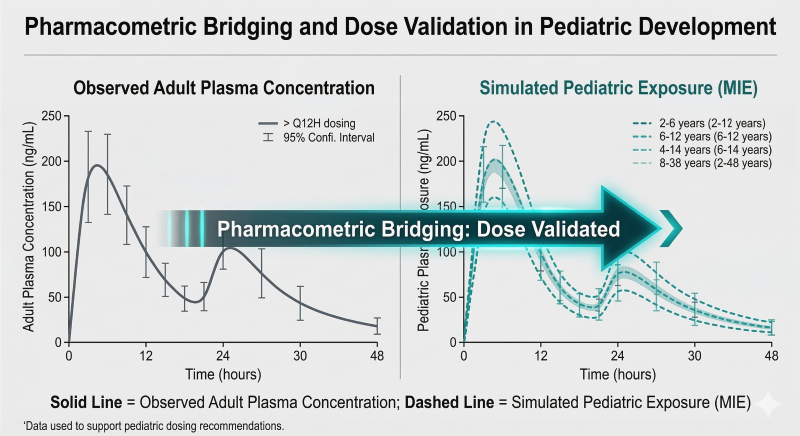 Figure 2: MIE Bridging. Using adult PK data to simulate and validate pediatric dosing without a new efficacy trial.
Figure 2: MIE Bridging. Using adult PK data to simulate and validate pediatric dosing without a new efficacy trial.
DDI Bridging and Organ Impairment
A new molecular entity used to require a dozen separate clinical pharmacology studies to look at interactions or the impact of renal impairment. Under the MIE paradigm, you run a single anchor study to validate the model and then simulate the remaining scenarios [1]. This virtual approach is now becoming the standard for oncology and rare disease dossiers.
Case Study: MIE in Action: Brexpiprazole (Rexulti)
One of the best examples of the transition to MIE is the pediatric dosing approval for Brexpiprazole. Instead of conducting a massive, multi year clinical efficacy trial in pediatric patients, the sponsor used a sophisticated Model Integrated Evidence approach [3].
The drug was already approved for adults, but the sponsor needed a label for patients aged 13 to 17 without a dedicated Phase 3 trial. They used a combination of adult efficacy data and a validated PopPK model to perform exposure response extrapolation. By proving that the drug exposure in adolescents matched the effective exposure in adults, they demonstrated pharmacological plausibility. The FDA accepted this as primary evidence of effectiveness, accelerating the label extension by about 18 months and saving tens of millions in clinical costs [3, 5].
4. The Evolution of Evidence Standards
| Feature | MIDD (The 2020 Standard) | MIE (The 2026 Standard) |
|---|---|---|
| Role of the Model | Supporting evidence for design | Primary evidence for the label |
| Clinical Requirement | Model must be confirmed by a trial | Model replaces a trial arm |
| Data Source | Small scale pilot studies | Large scale virtual twin simulations |
| Primary Goal | Dose selection | Regulatory biowaiver |
5. Closing the Validation Gap
To move from being informed to being integrated, the burden of proof shifts to the quality of the model itself. In 2026, reviewers look for three specific pillars [1, 2]. First is purpose fit validation, where the model is verified for the specific regulatory question. Second is uncertainty quantification, where you prove the model stays accurate even if assumptions vary. Finally, you need virtual twin heterogeneity, showing that your simulations account for real world diversity rather than just average volunteers.
 Figure 3: The Validation Framework. The three pillars required to move a model from supporting evidence to primary evidence.
Figure 3: The Validation Framework. The three pillars required to move a model from supporting evidence to primary evidence.
Conclusion: A New Era of Clinical Pharmacology
As the cost of clinical failure keeps rising, MIE has become the most powerful tool in the biotech toolkit. It is no longer a technical luxury, it is a commercial necessity. The leaders in 2026 will be the sponsors who can turn their data into a validated regulatory narrative that lets the math speak for itself [2]. At ClinPharm Dev Solutions, we specialize in building those bridges.
Is Your Data Working Hard Enough?
Stop running redundant trials. Let us help you turn your pharmacometric models into primary regulatory evidence.
Book a Strategic ConsultationAbbreviations
- DDI: Drug-Drug Interaction
- FDA: Food and Drug Administration
- MIDD: Model-Informed Drug Development
- MIE: Model-Integrated Evidence
- PBPK: Physiologically Based Pharmacokinetics
- PopPK: Population Pharmacokinetics
References
- FDA Guidance (2025). Model-Informed Drug Development: Integration of Modeling and Simulation into Regulatory Submissions.
- Marshall, S. et al. (2023). The Shift to Model-Integrated Evidence: Lessons from the MIDD Pilot Program. CPT: Pharmacometrics & Systems Pharmacology.
- FDA Summary Basis for Approval. Rexulti (Brexpiprazole) Pediatric Extrapolation Case Study.
- EMA Reflection Paper (2025). The Use of In Silico Modeling in the Tailored Clinical Approach for Orphan Biologics.
- Wang, Y. et al. (2024). Accelerating Pediatric Drug Development through Model-Integrated Evidence. Journal of Clinical Pharmacology.